Chapter 4: Advanced applications for identification and quantification of the phosphoproteome
Doi: 10.4155/FSEB2013.13.72
Authors
Robert Parker
Australian Proteome Analysis Facility (APAF)
Macquarie University
Sydney, Australia
e-mail: [email protected]
Mark P. Molloy
Australian Proteome Analysis Facility (APAF)
Macquarie University
Sydney, Australia
e-mail: [email protected]
About the Authors
Robert Parker
Robert Parker is a post-doctoral fellow at Macquarie University. His research interests cover the application of proteomic techniques to advance the understanding of biological phenomena. His current drive is to understand how aberrant kinase activity drives oncogenic transformation of cells in melanoma.
Mark P. Molloy
Mark Molloy is Associate Professor and Director of the Australian Proteome Analysis Facility at Macquarie University, Australia. His research interests are in biomedical uses of quantitative mass spectrometry in basic and translational research settings. A focus of his research is in the field of cancer research utilizing discovery and targeted mass spectrometry for biomarker applications and to investigate mechanisms of cell signaling.
Advanced applications for identification and quantification of the phosphoproteome
Introduction
Measuring global protein phosphorylation levels can be achieved using several methodologies. In this chapter, we present an up-to-date summary of the major techniques currently used by researchers, highlighting their applications in the study of emergent biological processes. With a strong focus on functional proteomics and mass spectrometry, we describe the latest application of this approach in combination with genetic and chemical manipulation.
The dynamic phosphoproteome
All lifeforms share the ability to respond to the environment that surrounds them. Signal transduction from receptors on the cell surface to the genome and other intracellular structures is the process by which environmental information is biochemically encoded and translated into an appropriate response. This process is exceptionally complex, with multi-dimensional information about extracellular goings-on forming intracellular networks within areas of confined spatial or temporal separation. Signaling networks form rapidly, coded mainly by rapid enzymatic post-translational modification (PTM) of proteins and bursts of small secondary messengers (e.g., Ca2+, cyclic-AMP, inositol phosphates and diacylglycerol). The best studied and an almost ubiquitous signaling mechanism is protein phosphorylation. Phosphorylation was first described in 1906 for the protein vitellin, however it took approximately 50 years before the first enzymatic phosphorylation of a protein was described, and the action of a specific group of ATPases known as protein kinases shown to be regulators of this process [1,2]. Today, after 50 years of further biochemical characterization and genome sequencing we have near complete knowledge of all 518 human protein kinases, and 160 phosphatases that regulate the phosphoproteome [2,3].
Protein kinases are responsible for the transfer of ɣ-phosphate from adenosine triphosphate (ATP) to specific hydroxyl-amino acids; serine, threonine and tyrosine in eukaryotes, with histidine, aspartic acid and arginine modification also prevalent in prokaryotes. Remarkable control of substrate specificity is demonstrated by kinases and phosphatases [4]. On average, human cells consist of 90% P-Ser, 10% P-Thr and 0.5% P-Tyr; however, these numbers do not reflect the natural abundance of these amino acids, rather a preference for serine and tight control over tyrosine phosphorylation. In counterbalance to the action of kinases, ~78 Ser/Thr protein phosphatases (PP), ~37 protein tyrosine phosphatases (PTP) and ~ 63 dual specificity phosphatases (DUSPs) catalyze the removal of phosphates (dephosphorylation) [2]. Reversible phosphorylation is limited by temporal and spatial aspects of gene expression. Often co-expressed and organized into discrete modules (cytoplasmic scaffolds, membrane microdomains and holoenzymes), the proximity of kinases and phosphatases to their substrates is tightly regulated to provide a high degree of specificity [5,6]. Beyond proximity control, structural constraints define the interaction of kinase/phosphatase with their substrates [4,7]. Many substrates share homologous primary sequences (consensus motifs) that flank P-sites and distal docking domains that provide optimal binding affinity.
The phosphoproteome is complex, and it is clear that the regulation of substrates by kinases and phosphatases is controlled by several biochemical factors to generate a system with relatively high specificity and control. Analysis of the phosphoproteome requires knowledge of the P-site location and concentration, and the activity hierarchy of each kinase or phosphatase in biological systems. In recent times, several technical hurdles have been overcome, allowing the large-scale identification of these features (see Boxes 1 and 2). Many techniques rely on established procedures updated with new chemical tagging or genetic tools, but in almost all cases these procedures have been revolutionized when combined with the latest methods for phosphopeptide enrichment and characterization by mass spectrometry (MS).
Defining kinase/phosphatase substrate relationships
Defining the enzyme–substrate relationships is a vital step to elucidate the phosphoproteome, and for kinases this can be readily achieved using the in vitro kinase assay. Classically, this approach requires the kinase of interest to be purified and incubated with the substrate(s) of interest, radioactive [32P]ATP and key co-factors essential for phosphorylation. If the kinase is able to phosphorylate the substrate, then [32P] is incorporated and detected by autoradiogram. This approach can be augmented with 1D and 2D gel electrophoresis or protein/peptide arrays to separate substrates, detected by phosphorimaging and often phosphopeptides can be sequenced by Edman degradation or MS (kinase substrate tracking and elucidation [KESTREL]) [8]. In general, the researcher can either screen lysates from biological samples or generate peptide libraries to define the consensus motif for the kinase under investigation. More recently, MS has been used to simultaneously detect and identify the phosphorylated peptide without the use of radioactivity. Liquid chromatography coupled to mass spectrometry (LC-MS) is a powerful tool in phosphoproteome analysis and most current methods utilize its superior ability to detect and identify thousands of phosphopeptides in a single analysis (see Box 1).
Whilst still highly useful, the in vitro kinase assay can suffer from false positive substrate identification owing to the lack of specificity achieved when purifying the kinase of interest. In a major advancement of this technique, genetic manipulation by site-directed mutagenesis can generate kinases with an analog-sensitive active site (AS kinase) [9,10]. AS kinases possess an active site specifically designed to utilize only a bulky synthetic analog of ATP that cannot be enzymatically processed by any native kinase. Detection of substrates can again be achieved classically with a [32]ATP analog or by MS. AS kinases are also highly sensitive to specific cell-permeable inhibitors, so these can be used to identify in vivo effects of kinase signaling.
Application: AS kinase to investigate cell cycle kinases
Quantitative MS has been utilized with AS kinase technology to investigate the cell cycle phosphoproteome [11,12]. The breakthrough use of this technology developed a chemical strategy able to purify thio-phosphate-tagged substrates with high purity from tryptic digests of the proteome. Using targeted hydrolysis of the phosphate diester with oxone, thio-phosphates can be isolated from natural thio-ester background formed by cysteine/methionine-containing peptides. In this work, Blethrow et al. demonstrated the effectiveness of their strategy in identifying low abundant substrates of Cdk1-cyclin B kinase in the whole cell and nuclear envelope of HeLa cells [11]. They validated many of the sites with in vivo phosphoproteomic datasets and carried out specific kinase assays for several of the substrates. Interestingly, this strategy revealed several substrates that did not correspond to the optimal Cdk1 consensus motif ([ST]Px[KR]), indicating that primary sequence motifs are not always adequate to assign substrates. A year later, a separate group utilized a quantitative approach to describe Cdk1 substrates in the budding yeast S. cerevisiae [13]. Again a cyclin-dependent kinase 1 (Cdk1) AS kinase mutant was generated however the AS-kinase specific inhibitor 1-NM-PP1 was used to block CDK1 phosphorylation. Using SILAC metabolic protein labelling, the authors investigated substrates from asynchronous and mitotic arrested cell populations after treatment with 1-NM-PP1. Thus, this group not only purified the substrates but also by screening for sites that reduced in quantity after inhibition in vivo substrates were proposed. Apart from mapping Cdk1 phosphorylation during the cell cycle, the authors mapped the evolution of phosphosites and observed few sites to be highly conserved with the majority found within disordered rapidly evolving regions. Interestingly, highly conserved sites are found within metabolic enzymes, indicating Cdk1 kinase-mediated regulation of metabolism to be ancient. For many sites, whilst the precise position was not conserved, there was a clustering of consensus sites within protein coding regions of genes from the same lineage. The authors conclude that the shifting of several P-sites may drive evolutionary plasticity of Cdk1 signaling in cell cycle and development, and they draw parallels with the evolution of transcriptional regulatory motifs.
Box 1: Next-generation proteomics.

Purification and separation of phosphopeptides for in-depth analysis of the phosphoproteome
Profiling the phosphoproteome in response to the chemical and genetic approaches discussed in this chapter requires in-depth analysis of all P-sites present in a given sample. For the majority of phosphoproteins, the percentage of occupied P-sites is usually far lower (usually sub-stoichiometry) than the detection sensitivity of today’s mass spectrometers. To overcome this, enrichment techniques are required to selectively purify phosphorylated proteins/peptides from the large proportion of background unphosphorylated proteins. Affinity purification of phosphopeptides is now routinely achieved with immuno-, metal oxide- and immobilized metal affinity-chromatography methods [15]. Antibodies work particularly well for the rare phospho-Tyr containing peptides or where specific kinase consensus motifs are known, and the aim is to enrich for substrates of a particular kinase. More general approaches use metal oxide, usually TiO2 or iron (Fe3+) immobilized metal ion chromatography (IMAC), however the deepest phosphoproteome coverage comes from combining enrichment techniques with chromatographic fractionation (e.g., strong cation/anion exchange (SCX/SAX) or hydrophilic interaction chromatography [HILIC]). Figure 2 demonstrates the steps in analysis of the phosphoproteome by SCX and metal ion chromatography. Initially, samples are lysed and proteins solubilized with detergents, critically phosphatase activity must be limited by adding inhibitors or denaturation with chaotropes and/or heat (Figure 2A). 5–10 mg of total protein is then digested in solution to peptides with trypsin, purified by solid-phase extraction and separated by a salt gradient on high-capacity SCX liquid chromatography (Figure 2B). Fractions are collected and phospho-peptides purified by metal ion affinity (Figure 2C). At a low pH, phosphopeptides remain negatively charged due to the low pKa whilst the majority of polar side chains in peptides will be positive or neutral. Under these conditions, stationary phases with immobilized metal chelating ions (E.g. Ti4+ or Fe3+) can form electrostatic interactions with the phosphonate group (PO42-) and efficiently purify phosphopeptides. Extensive washing is required to remove non-phosphopeptides, particularly those containing acidic amino acids, and high pH (>10) used to elute the bound phosphopeptides (Figure 2D). Both unbound and bound fractions are analyzed by LC-MS to allow a comparison of phosphoproteome to the proteome, and in some cases P-site occupancy can be calculated.
Application: Next generation proteomics provides in-depth phosphoproteome coverage
In mice, a large-scale analysis of nine tissues utilized 10 mg of protein extracted from each tissue and SCX-IMAC enrichment to separate and purify phosphopeptides has been performed [16]. From ~284,000 redundant phosphopeptide identifications, ~36,000 unique P-sites from 6300 different proteins were characterized. Striking differences in site occupancy occur for phosphoproteins in different tissues, and abundance of occupied sites does not reflect protein expression. These findings cement the importance phosphorylation networks play in cell differentiation to support the specialized function of tissues and organs. Phosphoproteomics of mitosis in HeLa cells identified ~20,000 phosphopeptides from 6027 proteins [17]. For the analysis of mitotic phosphorylation, the authors also calculated site occupancy for many of the sites identified, and they demonstrated that there is a large range of site occupancy during mitosis with some sites completely phosphorylated.
Box 2: Bioinformatics.

Combining in vitro and in vivo approaches
In recent years, several landmark papers have described the combined use of in vitro and in vivo approaches to define kinase-substrate relationships. The kinase rescue assay or kinase assay linked with phosphoproteomics (KALIP), requires phosphoproteomic data generated from untreated samples to be paired with two samples treated to knockdown (RNAi or specific kinase inhibitor) the activity of the kinase under investigation [22] (Figure 3A–E). There are several variations of the approach but all carry out an in vitro kinase assay on one of the kinase treated samples either at the protein or peptide level. All three samples are digested to peptides and subjected to phosphopeptide enrichment techniques, then samples are analyzed by quantitative MS to identify and determine the relative abundance of phosphopeptides. In vivo targets of the kinase are phosphopeptides that are reduced in quantity compared to control after in vivo knock-down experiment, but recovered by the in vitro kinase assay (Figure 3E2).
Application: KALIP Syk substrates in breast cancer
The KALIP strategy was first described in the detection of substrates of the spleen tyrosine kinase (Syk) in breast cancer [22]. Syk is known to act as an oncogene and tumor suppressor depending on cancer cell type and has diverse roles, from antigen signaling in adaptive immunity in B-cells to response to oxidative stress and cell differentiation. Using two cell lines, this work demonstrated that only ~5–19% of in vitro and in vivo sites were modulated in common. They confirmed several of these by performing immunoprecipitation and an in vitro Syk kinase assay detecting phosphorylation using an anti-phosphotyrosine antibody. Next, the authors generated a breast cancer cell line with a tetracycline inducible form of the Syk kinase and applying the KALIP approach identified 23 Syk substrates. Supporting their data, cortactin, a bona fide Syk substrate in breast cancer, was identified, whilst pathway analysis indicated substrates to be involved in immune signaling, cellular organization and centrosome function.
In vivo approaches to the phosphoproteome
The activity of kinases and phosphatases represents an important factor in understanding how the phosphoproteome is regulated in different cell states or phenotypes. Once in vitro associations have been described for substrate(s) and/or consensus motifs and specificity can be modelled, the next step is to confirm the activity of kinases and substrate phosphorylation in vivo. The ability to knock out/in genes in model organisms has been used widely to study the physiological functions of many proteins. For kinases and phosphatases, it is possible to generate gain of function or loss of function mutants, or implement functional knockouts using RNAi. These genetic screens offer an in vivo approach to disrupting kinase/phosphatase function and, combined with phosphopeptide enrichment and identification strategies, this will reveal functional consequences of kinase and phosphatase activity. However, several drawbacks exist in the use of genetic manipulations. RNAi is usually slow requiring 24–72 hrs effective knockdown; over this time scale signaling cascades will develop compensatory feedback loops, confounding results. Over long time-points or with any genetic manipulation, the influence of protein translation needs to be accounted for alongside secondary effects, including stress-induced pathways such as autophagy and apoptosis.
Application: In vivo dissection of mTOR signaling
Dissection of the TOR kinase complex (mTORC) has been achieved using quantitative phosphoproteomics [23]. mTORC signaling controls cellular growth and metabolism, and is often found perturbed in cancer, diabetes, obesity, cardiovascular disease, inflammation and neurodegeneration. Currently, TOR kinase is known to function with different roles in two distinct complexes, mTORC1 and mTORC2, which differ by their respective interaction with the proteins raptor and rictor. Phosphopeptides from mouse embryonic fibroblasts (MEF) with floxed gene silencing of raptor or rictor genes identified several downstream substrates of these complexes. The authors were able to differentiate, based on substrates, the differing functions mTORC1 and mTORC2 play in response to nutrients, growth factors and cellular energy. They identified S1859 in the carbmoyl-phosphate synthetase 2 (CAD) protein as a downstream component of mTORC1 signaling. CAD functions in the pyrimidine biosynthetic pathway required for DNA replication. Using motif analysis, small molecule inhibition and double knockout experiments, the authors identified S6-kinase linking mTORC to CAD phosphorylation. mTORC1-dependent phosphorylation of CAD on S1859 catalyzed its oligomerization and drove synthesis of pyrimidines and S-phase progression.
Application: RNAi of Tank binding kinase and tyrosine phosphatase Ptp61F
Alongside genomic screens, RNAi is extensively used to define the in vivo phosphoproteome. Recently, knock-down of the oncogene Tank-binding kinase-1 (TBK-1) identified signaling by Polo-like kinase 1 (PLK-1) [24]. 4621 peptides (~2000 protein) were identified by LC-MS of SILAC-labelled A549 cells. The group used the software MaxQuant to quantitate changes in phosphorylation, and mapped these to several signaling pathways. The substrates altered indicated that TBK-1 may drive progression through mitosis in lung cancer by activating PLK-1. However, RNAi is not only providing insight into the function of kinases, it has been used to extensively characterize the function of phosphatases. In SILAC-labelled drosophila cells, the non-transmembrane protein tyrosine-phosphatase (PTP) Ptp61F was knocked down with RNAi, and compared to rapid system-wide inhibition with a phosphatase inhibitor cocktail [25]. Over 10,000 phospho-sites were detected using an SCX-TiO2 workflow. The knockdown revealed a highly specific effect on the phosphoproteome regulating only 288 (4.4%) of the sites quantified. Amongst the regulated phospho-Y sites, several overlapped with previous studies using a PTP substrate trapping technique, several were novel and most likely direct substrates of Ptp61F. Four substrates were orthologs of substrates of the Abl/Bcr-Abl tyrosine kinase.
Pharmacological dissection of the phosphoproteome
In the search for pharmacologically valuable small molecules, thousands of kinase/phosphatase inhibitors are now commercially available, with many more under development. Several of these are already proving useful in the treatment of disease, with a plethora undergoing clinical trials [26]. For the direct study of the phosphoproteome, inhibitors represent a valid tool for unravelling the action of specific or a broad set of kinases/phosphatases. Despite the high degree of similarity between the ATPase active site of protein kinases, it has still been possible to design competitive inhibitors with a high degree of specificity. The melanoma drug vemurafenib is a highly specific inhibitor of the mutant MAP kinase protein B-raf. B-raf drives oncogenic transformation in several cancers (melanoma, thyroid, colon) and inhibition of mutant B-raf has demonstrated therapeutic potential [27]. Vemurafenib was designed by a combinatorial approach termed fragment-based lead design (FBLD), which uses small fragments of molecules to detect low-affinity compounds. These compounds are combined to produce highly specific high-affinity molecules. Apart from the obvious use in therapeutics, compounds designed this way can facilitate rapid functional knockout or be used as new affinity reagents to purify kinases/phosphatases [28]. Detecting the system level effects of small-molecule-based inhibition in vivo is key to understanding how patients respond to treatment. Phosphoproteomic procedures performed within model systems or tissue obtained from patients undergoing treatment with inhibitors have the potential to identify the biochemical response to treatment, revealing (a) disease related kinase substrates, (b) downstream signaling for drug detoxification, (c) activation of pathways involved in drug resistance, and (d) putative prognostic/predictive biomarkers. This type of metadata when associated with functional studies may facilitate more effective drug treatments through discovery of better dosing strategies to reduce development of resistance, non-specific toxicity and potentially identifying targets that may be suitable for combinatorial inhibition. Towards these aims, several in vivo phosphoproteomics studies have recently emerged that attempt to understand the relationship between drug sensitivity and pathway activation in cancer.
Application small molecule inhibition of kinases in cancer
Casado et al. described a label-free approach to demonstrate that quantitative analysis of the phosphoproteome can stratify cancer cell lines by tissue origin and sensitivity to different inhibitors [29]. The same group also showed that in acute myeloid leukemia (AML) activation of different signaling pathways in this cancer (PI3K/mTOR, MEK and JAK) leads to different levels of drug sensitivity in cell lines [30]. Utilizing phosphoproteomics and growth kinetics to correlate regulated P-sites these data indicate that classical markers for pathway activation could not always explain sensitivity to drug inhibition and resistance was often associated with parallel activation of other pathways (e.g., protein kinase C [PKC]). Together, their data indicates that not all cancer cells confer the classical single oncogene addicted status, and thus combined therapies will be more effective in controlling the proliferation of resistant tumor cell populations. Here, the strength of phosphoproteomics is to provide an unbiased account of pathways activated/deactivated in cancer and after kinase inhibition.
Conclusions
Phosphoproteomic analyses using MS are rapidly adding to our knowledge of the vast array of protein substrates in temporal and spatial cellular contexts. This has been achieved through development of robust enrichment and bioinformatic data analysis platforms. However, we are only at the beginning of cataloguing the phosphoproteome, with much yet to be done using a variety of model biological systems to establish a clearer picture of this detailed layer of cellular regulation. While proteomic techniques are now sufficiently advanced to generate this information, future challenges shift towards defining the function and regulation of individual phosphorylation sites on individual proteins in defined biological situations.
Executive summary
- Today, after 50 years of biochemical characterization and genome sequencing we have significant characterization of the 518 human protein kinases and 160 phosphatases that regulate the phosphoproteome.
- Defining the enzyme–substrate relationships is a vital step to elucidate the phosphoproteome.
- The way in which scientists identify and quantitate proteins has radically altered over the past 20 years with the implementation of modern MS.
- For the direct study of the phosphoproteome, inhibitors represent a valid tool for unravelling the action of specific or a broad set of kinases/phosphatases.
- Genetic screens offer an in vivo approach to disrupting kinase/phosphatase function and combined with phosphopeptide enrichment and identification strategies will reveal functional consequences of kinase and phosphatase activity.
- The vast amount of data generated by MS phosphoproteomic screens requires an informatics solution to all consecutive steps in data generation/processing and biological interpretation.
Key terms
Protein phosphorylation: The covalent attachment of a phosphate moiety (PO43-) to the hydroxyl side-group of certain amino acids. The reaction is facilitated by a large family of enzymes known as kinases. Each phosphate moiety generates an increase in ~80 Da to the overall mass, altering the local and global electrostatic properties leading to changes in protein function.
Kinase/phosphatase substrate relationship: The specificity by which phosphorylation is regulated. The identity of enzymes (kinases/phosphatases) responsible for control of phosphorylation at specific sites in substrate proteins.
Consensus sequence/motif: A conserved primary sequence of certain flanking amino acids surrounding a substrate phosphorylation site that is a recognition sequence for a given kinase.
Kinase inhibitor: Usually a small molecule developed as an ATP competitive inhibitor. Binds with different degrees of specificity to kinases and prevents activity by blocking ATP binding and hydrolysis within the enzyme active site.
References
- Fischer EH, Krebs EG. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J. Biol. Chem. 216(1), 121–132 (1955).
- Alonso A, Sasin J, Bottini N et al. Protein tyrosine phosphatases in the human genome. Cell 117(6), 699–711 (2004).
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 298(5600), 1912–1934 (2002).
- Ubersax JA, Ferrell JE, Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8(7), 530–541 (2007).
- Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. Scaffold proteins of MAP-kinase modules. Oncogene 26(22), 3185–3202 (2007).
- Tonks NK. Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J. 280(2), 346–378 (2013).
- Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell, 139(3), 468–484 (2009).
- Cohen P, Knebel A. KESTREL: a powerful method for identifying the physiological substrates of protein kinases. Biochem. J. 393(Pt 1), 1–6 (2006).
- Shah K, Liu Y, Deirmengian C, Shokat KM. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. PNAS 94(8), 3565–3570 (1997).
- Koch A, Hauf S. Strategies for the identification of kinase substrates using analog-sensitive kinases. Eur. J. Cell Biol. 89(2–3), 184–193 (2010).
- Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. PNAS 105(5), 1442–1447 (2008).
- Blasius M, Forment JV, Thakkar N, Wagner SA, Choudhary C, Jackson SP. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 12(8), R78 (2011).
- Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325(5948), 1682–1686 (2009).
- Altelaar AF, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14(1), 35–48 (2013).
- Beltran L, Cutillas PR. Advances in phosphopeptide enrichment techniques for phosphoproteomics. Amino Acids 43(3), 1009–1024 (2012).
- Huttlin EL, Jedrychowski MP, Elias JE et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143(7), 1174–1189 (2010).
- Olsen JV, Vermeulen M, Santamaria A et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3(104), ra3 (2010).
- Lemeer S, Heck AJ. The phosphoproteomics data explosion. Curr. Opin. Chem. Biol. 13(4), 414–420 (2009).
- Chou MF, Schwartz D. Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinformatics. Chapter 13, Unit 13 15–24 (2011).
- Linding R, Jensen LJ, Pasculescu A et al. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 36(Database issue), D695–699 (2008).
- Bennetzen MV, Cox J, Mann M, Andersen JS. PhosphoSiteAnalyzer: A Bioinformatic Platform for Deciphering Phospho Proteomes Using Kinase Predictions Retrieved from NetworKIN. J. Proteome Res. (2012).
- Xue L, Wang WH, Iliuk A et al. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. PNAS 109(15), 5615–5620 (2012).
- Kang SA, Pacold ME, Cervantes CL et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341(6144), 1236566 (2013).
- Kim JY, Welsh EA, Oguz U et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. PNAS 110(30), 12414–12419 (2013).
- Hilger M, Bonaldi T, Gnad F, Mann M. Systems-wide analysis of a phosphatase knock-down by quantitative proteomics and phosphoproteomics. Mol. Cell. Proteomics 8(8), 1908–1920 (2009).
- Christoffersen T, Guren TK, Spindler KL, Dahl O, Lonning PE, Gjertsen BT. Cancer therapy targeted at cellular signal transduction mechanisms: strategies, clinical results, and unresolved issues. Eur. J. Pharmacol. 625(1–3), 6–22 (2009).
- Chapman PB, Hauschild A, Robert C et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364(26), 2507–2516 (2011).
- Fang B, Haura EB, Smalley KS, Eschrich SA, Koomen JM. Methods for investigation of targeted kinase inhibitor therapy using chemical proteomics and phosphorylation profiling. Biochem. Pharmacol. 80(5), 739–747 (2010).
- Casado P, Alcolea MP, Iorio F et al. Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol. 14(4), R37 (2013).
- Alcolea MP, Casado P, Rodriguez-Prados JC, Vanhaesebroeck B, Cutillas PR. Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Mol. Cell. Proteomics 11(8), 453–466 (2012).
Figure 1: Strategies for phosphoproteomic quantitation. (A) Isotopic labels are incorporated metabolically or by chemical tagging and comparing the in spectrum ratio of phosphopeptides. (B) Label-free quantitation compares the intensity of phosphopeptides between different LC-MS analyses.
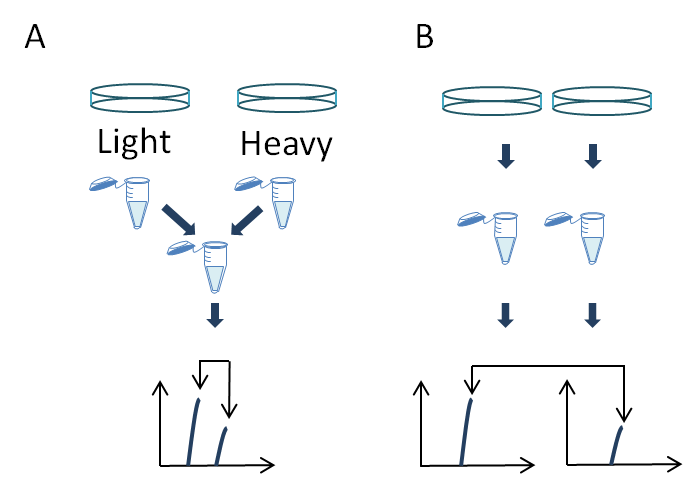
Figure 2: Phosphoproteomic analysis by combining strong cation exchange (SCX) and metal ion chromatography. (A) 5–10 mg of solubilized protein is digested to peptide by trypsin. (B) Peptides are separated by SCX chromatography. (C) Fractions are collected and phosphopeptides purified by interaction with metal chelating ions. (D) Purified phosphopeptides and unbound remainder peptides are analyzed by LC-MS.
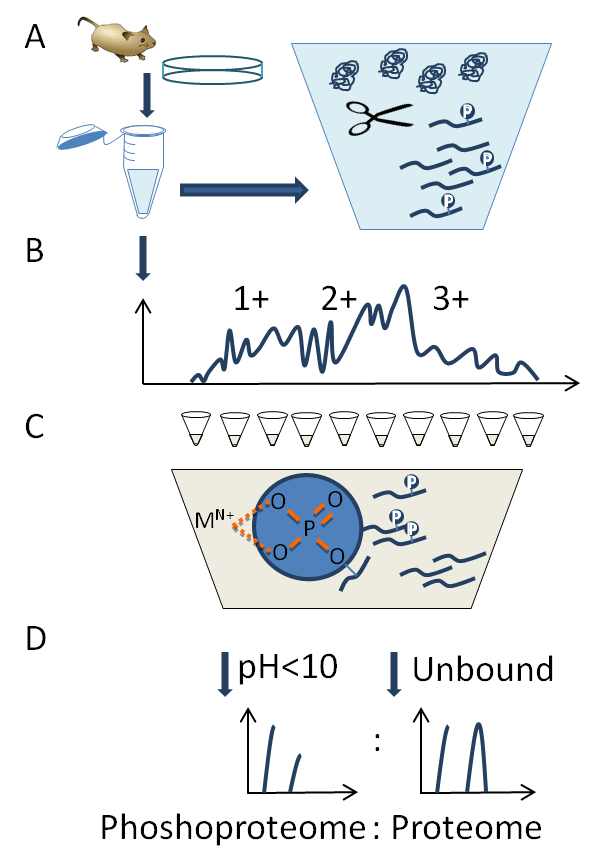
Figure 3: Kinase assay linked with phosphoproteomics (KALIP). (A) KALIP requires a quantitative approach and for several lab-grown model organisms (e.g., cells, flies, plants and mice) metabolic labelling using labelled amino acids (SILAC) or N15 media can be used to define three populations. (B) Specific in vivo kinase inhibition is carried out using RNAi or small molecule compounds in two samples to alter all substrates regulated by the kinase under investigation. (C) The samples are inactivated and proteins are digested to peptides with trypsin. (D) An in vitro kinase reaction is performed on one of the kinase-inhibited samples with a purified active version of the kinase inhibited in vivo. (E) Samples are mixed and phosphopeptides enriched and identified by MS, quantitative analysis allows specific ratios detected to define peptides regulated by kinase inhibition indirectly (1) versus direct kinase substrates (2) recovered by the kinase assay.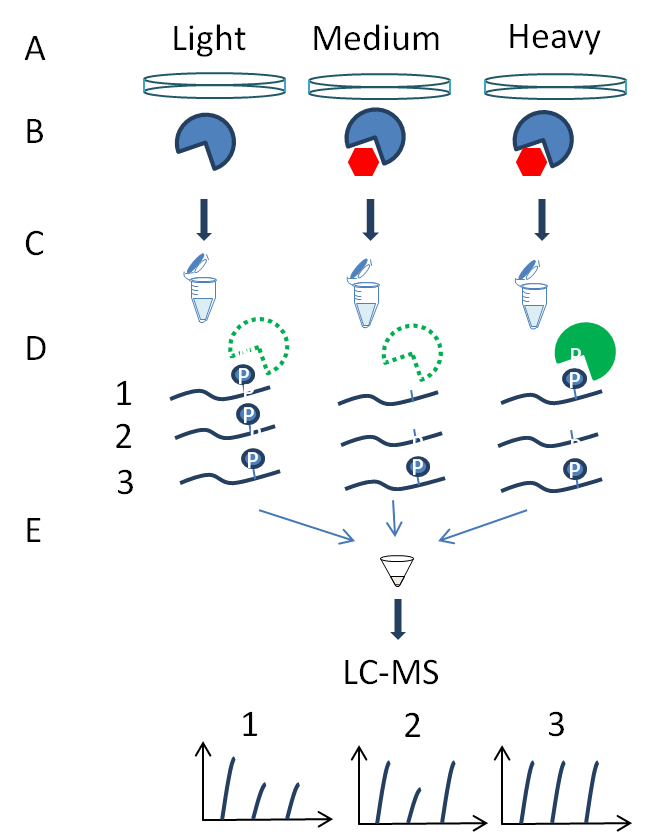
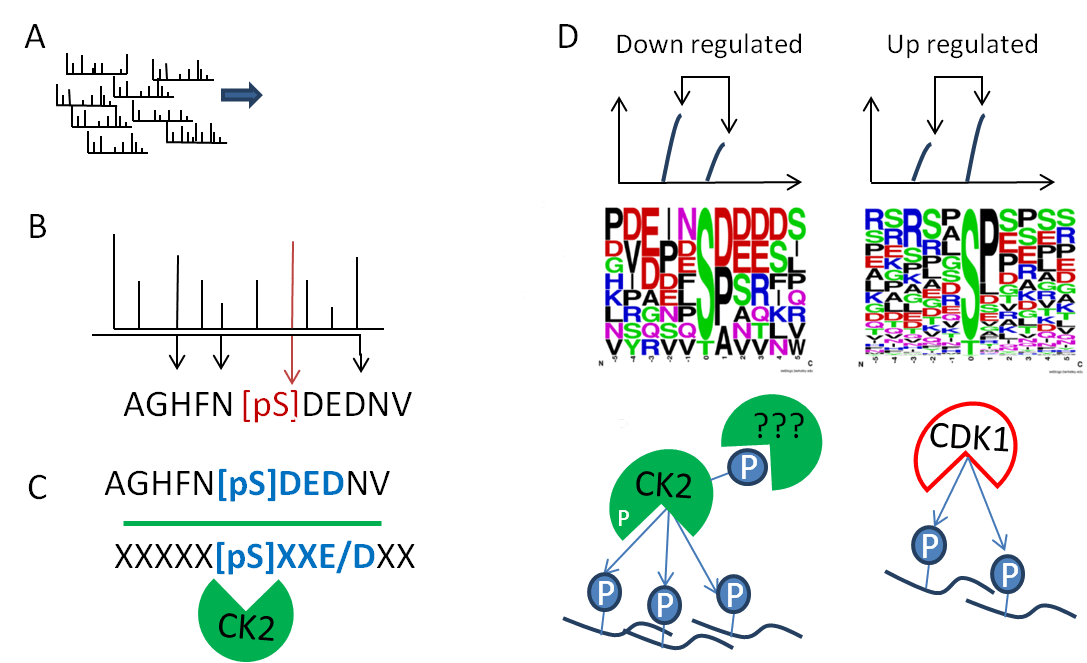
Figure 4: Bioinformatics. (A) Raw MS/MS spectra are processed in to peak lists. (B) Peptide sequence is identified by using protein database search tools and specific fragment ions provide site localization where possible. (C) Kinase–substrate relationships are identified by consensus motifs, contextual information and biochemical metadata. (E) Quantitation statistics are used to define regulated kinomes by substrate enrichment analysis and generate potential networks of kinase regulation (Weblogo generated at http://weblogo.berkeley.edu/).