Chapter 1: Automated sample preparation strategies for high-throughput analysis of body fluids
Doi: 10.4155/FSEB2013.13.77
Authors
Marco R. Bladergroen
André M. Deelder
Yuri E.M. van der Burgt
About the Authors
Marco R. Bladergroen
Marco Bladergroen obtained his Ph.D. in 2001 at the Institute of Molecular Plant Sciences, at Leiden University (The Netherlands). His thesis was entitled ‘Rhizobial secretion systems involved in avirulence’. Since then, he has worked as a senior scientist at the Center for Proteomics and Metabolomics at the Leiden University Medical Center (LUMC). His current research is mainly focused on the development of sample preparation procedures for high-throughput processing by means of automated liquid handling instruments. He has more than ten years of experience in this field. In addition he has gained experience on several aspects of proteomics and glycomics research.
André M. Deelder
André M. Deelder founded the Center for Proteomics and Metabolomics at the Leiden University Medical Center (LUMC). He obtained his masters in Biology (Zoology, Parasitology) at Leiden University in 1971. In 1973 he obtained his Ph.D. at Leiden University researching the immunology of helminth infections. From 1973 to 1978 he performed research on quantitative immunofluorescence and immunoenzyme techniques for diagnosis of schistosomiasis. In 1978 he became associate professor and group leader of the schistosomiasis group. In 1985 he was appointed a full professor, retiring in 2014. His research has focused on the immunology and epidemiology of schistosomiasis, the glycobiology of schistosomiasis and the structural and functional studies of parasite glycoconjugates. During these studies he pioneered the development of anti-glycan monoclonal antibody panels for detection of schistosome circulating antigens, resulting in a large number of internationally funded collaborative research projects. In his role as a chairman of the research division of the LUMC he set up a biomolecular mass spectrometry group with an infrastructure which is – in the field of clinical proteomics and metabolomics – unique for The Netherlands in both size and scope. This group has evolved into an independent department, recently named the Center for Proteomics and Metabolomics.
Yuri E.M. van der Burgt
Yuri E.M. van der Burgt obtained his Ph.D. in bio-organic chemistry at Utrecht University (The Netherlands) in 1999. At the Leiden University Medical Center (LUMC) he started his work in the new field of MS-based proteomics and contributed to the establishment of a glycoproteomics facility. After his postdoc he worked for three years as a scientist at the biotechnology company Crucell (The Netherlands), where he was responsible for the analysis of recombinant proteins. To strengthen his expertise in the field of biomolecular MS he moved to the FOM-Institute in Amsterdam to work on biomarker discovery within the Netherlands Proteomics Center (NPC). Then, to follow his ambitions in clinical research he returned to the LUMC as an associate professor at the Center for Proteomics and Metabolomics. He is responsible for multiple MS-platforms that are applied both for in-depth analyses and high-throughput clinical proteomics studies. In collaboration with the department of Clinical Chemistry a selection of the developed methods are currently transferred into routine assays.
Automated sample preparation strategies for high-throughput analysis of body fluids
Abstract
Sample preparation is an important step for LC-MS-based peptide and protein analysis because of the high complexity of biological specimens. The preparation methods should be accurate, precise and robust, and aimed at the research question to avoid the presence or introduction of unwanted or interfering substances. Previously, we have established fully automated sample preparation pipelines that allow high-throughput MS-profiling of body fluids. These workflows can also be applied to LC‑MS‑based proteomics experiments. In this chapter, protein digestion, solid-phase extraction and affinity purification are outlined, and considerations prior to sample preparation are proposed.
Introduction
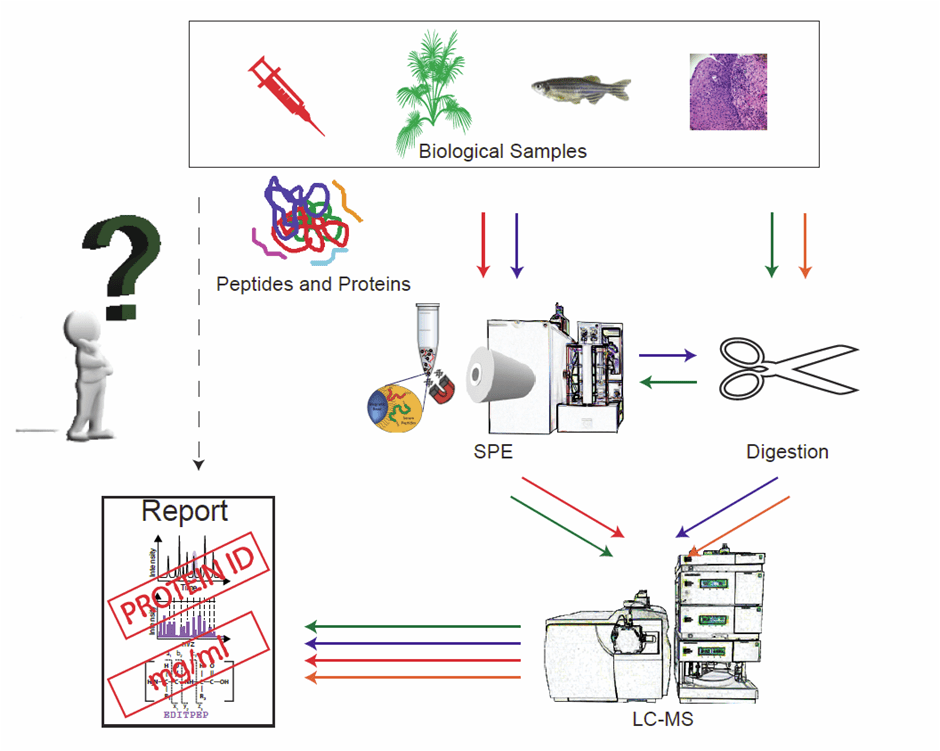
Sample preparation of biological specimens is an essential part of LC-MS-based proteomics because of the inherent complexity in terms of variety in components and their concentrations. Various strategies have been developed in order to obtain sample (protein) fractions for further identification and quantification by mass spectrometry. The route for sample prep that one will follow obviously relates to the actual research question taking into account that any bias should be avoided (Figure 1). For comprehensive coverage of the proteome, the isolation of as many peptides and/or proteins as possible is required, whereas in other cases only capture of a small set of peptides is required to answer a very specific question. In our laboratory, we have developed fully automated sample preparation pipelines that allow MS profiling of body fluids at high-throughput (500–1000 samples per 24 hours). Initially, most of these profiles were acquired on matrix-assisted laser desorption ionization (MALDI) time-of-flight (TOF) mass spectrometers, and this combination is still extremely powerful in terms of low costs and short analysis times. Moreover, the sample preparation workflows for profiling purposes are also compatible with LC-MS read-outs. Some basic requirements and techniques in high-throughput sample preparation protocols are:
- Standardized sample collection
- Protein digestion
- Solid-phase extraction (SPE)
- (Immuno-)affinity purification and depletion
Increasingly, and in particular for diseased cohort studies, high-throughput analysis is required, i.e. short measurement times or multiplexed analysis. High-throughput platforms that allow the analysis of large numbers of samples are needed in both research and hospital settings, where doctors and patients demand fast answers. Efforts to optimize throughput parameters should therefore be considered during the design of a procedure. Furthermore, when performing quantification experiments it is important to reduce the number of handling steps prior to analysis, in order to obtain minimal loss of analyte, and to have a good estimation or, better, measurement of the efficiency of recovery. A third issue to take into account is the nature of the sample. A short survey on the ‘Web of Knowledge’ with the search term ‘proteomics’ reveals almost 125,000 contributions (December 2015). Combining this with the search term ‘human’ results in more than 46,000 papers, of which about half are cancer-related. The majority of these studies were based on blood, serum and/or plasma samples. This is probably because this sample is relatively easy to obtain, the concentration of protein/peptides in “blood-derived” samples is high (reference range for serum is 60–85 g/l total protein) and it is generally assumed that blood reflects the state of the body. With regard to other body fluids, although urine can be obtained non-invasively, it is much less used in proteomics (approximately 1,000 publications in a similar search). This is probably because the concentration of proteins and peptides is much lower in urine (normal total protein concentrations of about 20 mg/l in a standardized sample based on healthy individuals [1]) compared to plasma and serum, the presence of high salt concentrations, thereby obstructing sample preparation for LC-MS, and importantly the proteins and peptides present in urine do not correlate with the state of the body to a similar extent as blood samples do, with the exception of, for example, kidney diseases. It should be noted that in the case of urine, samples contain large amounts of salts that require some modification of preparation protocols for serum or plasma. Finally, cerebrospinal fluid (CSF) is a relatively “clean” sample that can often be further used without extensive preparation steps; however, it cannot be easily obtained and only in small volumes. In this chapter, various examples of sample preparation techniques will be highlighted that can be combined with bottom-up as well as top-down proteomics workflows. Although initially developed for high-throughput profiling, these sample preparation strategies are very well suited to further LC-MS analysis ([2] and references therein). This being said, we also acknowledge that this overview cannot describe the entire field of reported sample preparation routes.
Figure 1: Decision tree to go from sample to report. Four routes are shown, via solid-phase extraction (SPE) and/or protein digestion, with LC-MS read-out.
Standardized sample collection and storage protocols are crucial for appropriate sample preparation. The aim is to time-snap the state of the sample, or in other words, to keep the composition and quality of the sample as close as possible to the time point at which the sample was taken. In the case of serum and plasma, important parameters are processing times (e.g., the clotting time of a blood sample), storage times, storage temperatures and number of freeze–thaw cycles [3] (Box 1). Obviously, the choice of sample type itself defines later sample preparation. For plasma samples, additives such as heparin or citrate are needed to prevent clotting. Heparin is reported to influence protein chromatographic properties as a result of negative charges, as well as the anti-coagulant ethylenediaminetetraacetic acid (EDTA). This effect has to be taken into account in the analysis or these additives need to be removed prior to analysis. Clotting factors are removed in serum samples, however proteins or peptides with affinity to these clotting factors can be removed as well and thereby influence the sample composition. Clotting time and incubation time before removal of blood cells also determine the resulting sample (reviewed in [4]). Another variable that is of great importance in sample preparation is the consumables that are used for collection and processing. It is known that certain brands of plastic containers release more polymers during sample processing than others [Bladergroen MR, Unpublished data, 5]. For example, we detected bovine serum albumin (BSA) from adhesive foils used to close microtitration plates as a major interfering compound. Proteins and peptides may also adhere to tube walls, giving rise to significant differences in protein composition between two aliquots of the same sample processed in different tubes [6,7].
Protein digestion
Protein digestion, either enzymatically or chemically, is included in almost any sample preparation protocol aiming for MS-based protein identification and quantification. This approach is generally referred to as shotgun or bottom-up proteomics [8,2]. A crude sample may contain thousands of different types of proteins, with large variety in concentration, and digestion will further increase sample complexity. In the early days of MS-based proteomics, sample complexity was reduced prior to digestion by gel-based methods. Protein separation by two-dimensional (2D) gel electrophoresis may result in the isolation of single proteins, but subsequent digestion and the analysis (or identification) of each protein (or protein fraction) via a peptide mass fingerprinting approach is a tedious job and the analysis of one single biological sample will take a long time. Therefore, this separation method is not an option for high-throughput analysis. As an alternative, 1D-gel separation is often applied to yield approximately a dozen fractions, or alternatively by preparative (2D)-LC approaches. These methods provide a convenient number of fractions to allow identification through LC-MS/MS workflows. It should be further noted that in both gel-based sample fractionations, not all proteins will enter the gel, and therefore these proteins are lost for analysis. Liquid-based fractionations, such as 2D-LC or liquid–liquid extraction, could overcome such a loss. In the case when no fractionation is performed at all obviously no material is lost, but at the cost of decreasing performance of single LC methods for peptide or protein separation [9]. In other words, even while digestion is a non-targeted approach to analyze as many peptides/proteins as possible simultaneously, sample loss will always occur and for the comprehensive analysis of a proteome one will need to perform multiple experiments, each aimed at a different fraction.
Once a choice has been made regarding the pre-fractionation technique, the sample or fraction of sample is subjected to proteolytic or chemical digestion (proteolysis) leading to a set of peptides that can be analyzed by LC-MS. Several factors influencing the completeness of digestion play a role, as was recently reviewed [10]. The most obvious parameter in proteolysis is the selection of the protease itself. In addition, it can be beneficial to combine multiple proteases or to combine bottom-up data from various digestions. Next, the solubility of the protein and access of the proteolytic sites are important. One (extreme) example of this phenomenon accounts for membrane proteins. These proteins are intrinsically poorly soluble in the aqueous conditions that are often used for enzymatic digestion. Addition of organic solvents may improve the solubility of these proteins, but addition of too much organic solvent will reduce the activity of the proteolytic enzyme. The analysis of the membrane proteome is an art in itself and is discussed in more detail in another chapter of this book, as well as in a recent publication [11]. Addition of chaotropic agents or surfactants may also assist in the denaturation of proteins to give better access to the cleavage sites, but some of these agents are not compatible with MS and are difficult, if not impossible, to remove after digestion (for instance sodium dodecyl sulfate (SDS)). Other denaturing compounds that are more compatible with MS or better to remove are not equally strong in their denaturing capacity. Proc and co-workers performed an elegant and fairly extensive study on the effect of such compounds, including several solvents, on the quantitative digestion in time of a series of predefined proteins [12]. They concluded that there is not a single digestion protocol that is suitable for all proteins to be digested with 100% efficiency. Nonetheless, sodium deoxycholate (DOC) in combination with a 9-hour digestion time gave the most stable results (~80% digestion efficiency) with the highest average reproducibility. Still, examples exists (e.g., alpha-1-acid glycoprotein 1) where SDS outperforms DOC, but as stated earlier, SDS should be avoided if possible. The effect of these agents also depends on the proteolytic enzyme. While guanidine inhibits the enzyme activity of trypsin, this denaturing agent is, up to a 2 M concentration, perfectly suitable for digestion by LysC [13]. A second factor influencing the completeness of digestion is the digestion time. Generally, an overnight incubation is performed for digestions with trypsin; however, such long incubation times hamper high throughput. Previously, it was shown that for a certain group of proteins, digestion reached a maximum after only 4 hours, whereas for another group of proteins the maximum was not even achieved after 16 hours of digestion [12]. Too long digestion times are also not good, as several proteins showed a reduction in detection efficiency after their optimal incubation time, possibly owing to chemical modification or degradation. Furthermore, one should keep in mind that, as the authors state: ‘…digestion may reach completion, but the digestion is still not complete…’. The research question should determine if this is significant or not. For studies where identification is the main goal, this might be sufficient, while for an absolute quantitative analysis this obviously is not the case. In the latter case, precise knowledge on the extent and reproducibility of the digestion is of utmost importance. The degree of completeness can be quantified by following the digestion of an elongated version of a stable-isotope-labelled internal standard [14], or by monitoring multiple tryptic peptides from the same protein [15]. Moreover, it has been demonstrated that the source or batch of trypsin can determine the outcome, as was recently overviewed [16]. As a final remark, several methods have been described to shorten incubation times to minutes, for instance the guanidine-HCl/LysC combination described by Poulsen and co-workers, as mentioned earlier [13], and pressure-aided digestion [17], or even to seconds as demonstrated in temperature elevated or microwave-assisted digestion on trypsin-coated magnetic beads [18,19].
A solution to the loss of peptides owing to post-digestion sample clean-up, generally performed by reversed-phase (C18) tip-based solid-phase extraction (SPE), could be the online coupling of the digestion step with LC-MS by means of in-column digestion, often referred to as immobilized enzyme reactors (IMERs) [20]. The proteolytic enzyme is bound to a solid support and packed in a column. Reaction times may decrease to as low as 77 seconds and the resulting peptides can be measured online on an LC-MS platform [21]. In a recent publication, the use of a continuous-flow IMER at elevated temperatures is described. Application of this method reduced the analysis time not only as a result of the increased trypsin concentration but also by eliminating the need for reduction and alkylation. Moreover, the number of identified proteins increased by approximately 30% [22]. Two other recently developed digestion-derived methods, the so-called ‘pseudoshotgun’ (PSG) method and filter-aided sample preparation (FASP) [23], resulted in increased sensitivity owing to reduced loss of peptides. The first method is a 1D-gel based method with short (15 minutes) separation times. After the (partial) gel separation, the gel is sliced, then each gel-slice is subjected to enzymatic digestion and the resulting peptides are pooled again for subsequent LC-MS analysis. As mentioned earlier, SDS is detrimental to MS and requires removal prior to analysis, although still considered to be the most efficient detergent to solubilize proteins. Since SDS binds strongly to proteins, only a few methods have been reported that successfully remove this detergent. With FASP, digestion is directly performed on a molecular cut-off filter after removal of SDS through filtration. With these methods, it was possible to analyze the proteome of 250 melanoma cells with an identification rate higher than with conventional methods [24].
Solid-phase extraction
A second sample preparation technique that is widely used in MS-based proteomics is solid-phase extraction (SPE). With SPE, compounds are isolated on basis of their chemical and physical properties, which determine their distribution between a mobile liquid phase and a solid stationary phase. After binding of the molecules with the correct properties, the remaining compounds are washed away and the bound molecules are eluted from the solid phase by changing the mobile phase into the elution solvent. In the case of chromatography, an (analytical) column is used multiple times to allow separation of compounds by elution with a gradient, while in the case of SPE, the cartridge (or column) is usually disposed after each sample and no gradient is applied for elution (one-step elution). Thus, in theory, all compounds present in the sample are captured with chromatography, while with SPE only a certain group of analytes is isolated, depending on the solid phase. Therefore, SPE is mainly used to clean up a sample and reduce sample complexity. For protein analysis with MS, it is often used to remove salts and other impurities that might cause ion suppression. However, by carefully choosing the right SPE sorbent, higher selectivity can be achieved. This selectivity also means that in principle with SPE only part of the sample is analyzed, because not all compounds will be isolated, but only those compounds that match the binding capabilities of the sorbent. SPE material is available in various formats, including columns, cartridges, plates, micropipette tips and functionalized magnetic beads. Magnetic beads have several advantages over the other formats. First, their small size allows a higher concentration rate and therefore sensitivity owing to their large binding surface area [25,26]. Second, the use of magnetic beads is easily automatable, providing a highly consistent high-throughput method, although a high‑throughput method has also been shown for SPE with cartridges [27]. Recently, an automated method for tip-based SPE has been developed [28]. However, our personal experience with these tips is that the SPE material itself can easily act as a blockage in the tip, requiring under and overpressures in the tip during aspiration and dispense, which is hard to achieve with robotic pipetting systems, resulting in uncontrolled aspirated and dispensed volumes [Bladergroen MR, Unpublished Data].
The most commonly used SPE materials in proteomics are reversed phase for the isolation of proteins and peptides, normal phase including HILIC for the isolation of glycosylated proteins and peptides, and ion exchange material for the isolation of charged peptides. More SPE materials are displayed in Table 1. A fairly extensive overview is also given in a review by the group of Ole Jensen [29]. Recent advances in ion-exchange chromatography have been made with mixed-bed materials. Mommen and co-workers combined weak anion exchange (WAX) with strong cation exchange (SCX) material [30]. In this way, they were able to perform a salt-free elution using a diluted formic acid solution to which DMSO was added, while optimizing the WAX/SCX ratio. Peptide recoveries were significantly improved in comparison to SCX alone with a WAX/SCX ratio of 4. This improved recovery could be explained by the so-called Donnan effect, as well as by the absence of salts (that are usually incompatible with MS) by using a pH block-gradient [31]. This Donnan effect is also highly responsible for the binding of proteins to newly developed core-shell nanoparticles. Core-shell nanoparticles are composed of a solid interface covered with a shell of charged polymers either as microgel or as densely grafted linear polymers (so-called polyelectrolyte brushes). The exact mechanism that is involved in the binding of proteins to these nanoparticles is described in detail in ref. [32]. Such nanoparticles have successfully been applied in the isolation of low-abundance proteins, as not only electrostatic interactions are important, but these nanoparticles also have molecular sieving properties, which prevent large high-abundant proteins such as BSA from being captured. Further advantageous properties of these nanoparticles are their concentrating capacity, large surface area and their protectiveness against enzymatic degradation and other environmental influences on the captures peptides [33].
Affinity purification
Affinity purification is based on the reversible and non-covalent interaction of the protein or peptide of interest with naturally occurring (or synthetic analogues of) biomolecules, such as other proteins, lectins, nucleotides or dyes. Affinity purification is a popular chromatographic method for the purification of compounds in larger quantities, for instance immunoglobulins (IgG’s) with protA or protG. It is however also a convenient method for SPE of smaller sample volumes. As presented in the previous section and in Table 1, many chromatographic materials for affinity purification are available [34]. These can be either used for enrichment or depletion. When used for enrichment, in general more information about the analyte of investigation is already available and the research question has focused to a very specific group of proteins or even a single protein. When used for depletion, the aim is to remove or at least to reduce the high-abundance proteins to allow access to the low-abundance ones (“in-depth analysis”). For the latter application, products that make use of a mix of antibody-based affinity material are commercially available, such as the Proteoprep 20 Plasma immunodepletion kit from Sigma, which removes the 20 most abundant proteins in a single purification step [35], or MARS from Agilent [36]. Qian and co-workers used the commercially available ProteomeLab™ IgY12 affinity column (Beckman Coulter, Fullerton, CA) in combination with a custom-made Supermix column to separate more than 50 high- and medium-abundance proteins from the lower-abundance ones in a plasma sample [37]. A nice example of immuno-affinity (IA) enrichment was shown by Halfinger and co-workers [38], which involved a thermal protein precipitation step, followed by liquid–liquid precipitation before IA chromatography to quantify the low-abundance heart failure marker N-terminal pro-B-type natriuretic peptide (NT-proBNP). This example showed a combined preparation method to deplete high-abundance proteins followed by concentration of a specific low-abundance protein through immuno-affinity. The simple and cost-effective method could be used to unmask low-abundance proteins in general. Finally, it should be noted that another important and widely used application of affinity purification concerns the isolation of phosphoproteins through metal-affinity SPE. In addition, the isolation of glyco-proteins or -peptides through lectin-affinity is a field of its own. These subjects are however outside the scope of this chapter; reference [39] and references cited herein are suggested for further reading.
SISCAPA, a combination of digestion, SPE and affinity capture
A combination of protein digestion, SPE and affinity capture is used in the so-called SISCAPA method, which stands for stable isotope standards with capture by anti-peptide antibodies. This targeted protein quantification approach was first reported in 2004 by Anderson and co-workers [40] and comprises the isolation of a known peptide (from a biomarker protein) by means of antibody coated SPE-material after trypsin digestion of the total sample proteome. Where protein digestion was originally used in the discovery stage of MS-based proteomics studies to allow protein identifications, SISCAPA aims for absolute quantification in a clinical setting. This quantification relies on the addition of known amounts of a stable-isotope-labelled peptide (with the same amino acid sequence) to enable determination of the concentration of the endogenous peptide and thus the endogenous protein with high precision (CV < 5%). As discussed earlier in this chapter, for this purpose the extent and reproducibility of the digestion should be determined since the digestion often does not run into completion.
A number of immunoaffinity techniques in combination with MS have been reviewed earlier [41]. Although ELISA still outperforms MS-based techniques with regard to sensitivity, the potential of SISCAPA to allow quantification of low‑abundant proteins in body fluids is generally acknowledged. Further increases in sensitivity are expected from improvements in MS equipment. Recently, the SISCAPA workflow has been automated by means of magnetic beads and concurrently improved sensitivity to the pg/ml range by increasing the sample volumes [42]. Similar methodologies have been proposed based on immunocapture at the protein level [41]. Here, the wider availability of antibodies against many proteins could be advantageous. It is further suggested that sensitivity improves with the number of so-called proofreading steps [43]. A proofreading step is regarded as a step in an assay that results in a stepwise decrease in the error fraction. We foresee that the combination of the two techniques mentioned by Ackermann and Berna [41] will add extra proofreading steps (two different antibodies instead of one) and together with the improvements in SISCAPA workflows will increase sensitivity even more.
Relevance of automation
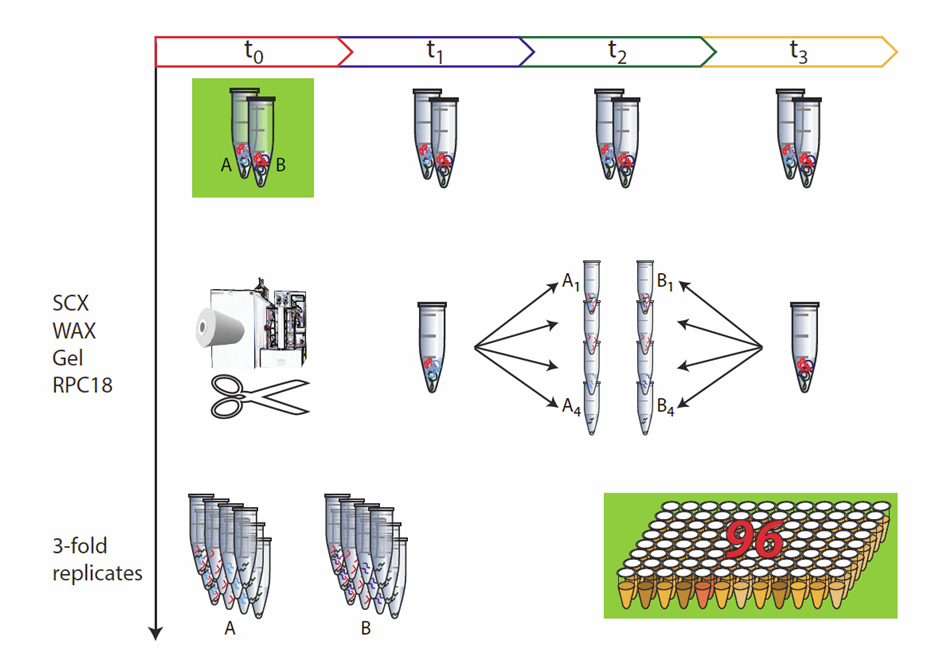
Sample throughput and robustness of the analytical workflow are of great importance in most types of applications, starting with biological specimens. In MS-based proteomics, sample throughput has increased enormously as a result of developments in automation, MS technology and improved speed in data handling and processing. In order to balance the speed of analytical sample preparation procedures with MS-based proteomics, the application of high-throughput robotic systems for sample workup is essential. Furthermore, processing of a relatively small number of samples can rapidly lead to a large number of samples for MS measurements by fractionation, purification steps and replicates, as exemplified in Figure 2. In addition to increased sample throughput, automation improves the accuracy and precision of the workflow. Provided a liquid handling system is calibrated and maintained on a regular basis, such an automated platform will perform each procedure in a standardized way over time, whereas larger variation is introduced when the same procedures are carried out by different people.
Figure 2: Relevance of automation: not only large cohorts require high-throughput strategies. With a relatively easy experimental setup such as three replicates for a given specimen, sampled at four different time points, processed by four separation strategies, the number of samples to analyze increases rapidly.
Executive Summary:
- A good experimental design involves rational selection of sample collection protocols.
- Two widely used sample preparation techniques in MS-based proteomics are protein digestion and solid-phase extraction (SPE). Proteolysis yields peptides that are suitable for identification purposes and SPE is applied to reduce sample complexity and to increase protein or peptide concentrations.
- SPE comes in many forms and with a wide variety of chemical principles.
- For absolute protein quantitation, it is important to monitor the completeness of the digestion process.
- The recent introduction of a combination of digestion, SPE and affinity capture (SISCAPA) allows absolute protein quantitation.
- Automation is of key importance to allow throughput of a high number of samples in a reasonable timeframe, in a robust and standardized workflow, providing precise, reproducible and accurate data.
Key Terms:
Cartridges for SPE: Small (disposable) columns that can be applied for isolation of peptides and proteins in an automated fashion. Both loading and elution (extraction) can be performed at high pressure in an automated way by using a cartridge exchanger device.
Functionalized Magnetic Beads: Uniform and disposable paramagnetic monodisperse microparticles (µm) with a specific and defined surface for the adsorption or coupling of molecules such as peptides and proteins. Magnetic bead chromatography is suitable for automation of sample preparation procedures.
Proteolysis: The breakdown of a protein into smaller peptides through enzymatic or chemical digestion. Enzymes (or proteases) with robust specificity commonly used in MS-based proteomics are trypsin, chymotrypsin and LysC. Chemical degradation is less specific and mostly occurs through acid hydrolysis.
Accuracy, precision, and reproducibility: Accuracy is the closeness of the measured result to the true value, precision (or repeatability) is the closeness of the results from multiple measurements (degree of scatter), reproducibility expresses all intra-laboratory variations, or the precision between different laboratories.
References
- Mischak H, Kolch W, Aivaliotis M et al. Comprehensive human urine standards for comparability and standardization in clinical proteome analysis. Proteomics Clin. Appl. 4(4), 464–478 (2010).
- Nilsson T, Mann M, Aebersold R, Yates JR, III, Bairoch A, Bergeron JJ. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 7(9), 681–685 (2010).
- de Noo ME, Tollenaar RA, Ozalp A et al. Reliability of human serum protein profiles generated with C8 magnetic beads assisted MALDI-TOF mass spectrometry. Anal. Chem. 77(22), 7232–7241 (2005).
- Lista S, Faltraco F, Hampel H. Biological and methodical challenges of blood-based proteomics in the field of neurological research. Prog. Neurobiol. 101–102, 18–34 (2013).
- Drake SK, Bowen RA, Remaley AT, Hortin GL. Potential interferences from blood collection tubes in mass spectrometric analyses of serum polypeptides. Clin. Chem. 50(12), 2398–2401 (2004).
- Hsieh SY, Chen RK, Pan YH, Lee HL. Systematical evaluation of the effects of sample collection procedures on low-molecular-weight serum/plasma proteome profiling. Proteomics 6(10), 3189–3198 (2006).
- Villanueva J, Philip J, Chaparro CA et al. Correcting common errors in identifying cancer-specific serum peptide signatures. J. Proteome Res. 4(4), 1060–1072 (2005).
- Hunt DF, Yates JR, III, Shabanowitz J, Winston S, Hauer CR. Protein sequencing by tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A 83(17), 6233–6237 (1986).
- Cox J , Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 80, 273–299 (2011).
- Switzar L, Giera M, Niessen WM. Protein digestion: an overview of the available techniques and recent developments. J. Proteome Res. 12(3), 1067–1077 (2013).
- Vuckovic D, Dagley LF, Purcell AW, Emili A. Membrane proteomics by high performance liquid chromatography-tandem mass spectrometry: Analytical approaches and challenges. Proteomics 13(3–4), 404–423 (2013).
- Proc JL, Kuzyk MA, Hardie DB et al. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 9(10), 5422–5437 (2010).
- Poulsen JW, Madsen CT, Young C, Poulsen FM, Nielsen ML. Using guanidine-hydrochloride for fast and efficient protein digestion and single-step affinity-purification mass spectrometry. J. Proteome Res. 12(2), 1020–1030 (2013).
- Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2(8), 587–589 (2005).
- van den Broek I, Smit NP, Romijn FP et al. Evaluation of Interspecimen trypsin digestion efficiency prior to multiple reaction monitoring-based absolute protein quantification with native protein calibrators. J. Proteome Res. (2013).
- Vandermarliere E, Mueller M, Martens L. Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom. Rev. 32(6), 453–465 (2013).
- Yang HJ, Hong J, Lee S, Shin S, Kim J, Kim J. Pressure-assisted tryptic digestion using a syringe. Rapid Commun. Mass Spectrom. 24(7), 901–908 (2010).
- Jeng J, Lin MF, Cheng FY, Yeh CS, Shiea J. Using high-concentration trypsin-immobilized magnetic nanoparticles for rapid in situ protein digestion at elevated temperature. Rapid Commun. Mass Spectrom. 21(18), 3060–3068 (2007).
- Lin S, Yao G, Qi D et al. Fast and efficient proteolysis by microwave-assisted protein digestion using trypsin-immobilized magnetic silica microspheres. Anal. Chem. 80(10), 3655–3665 (2008).
- Yamaguchi H , Miyazaki M. Enzyme-immobilized reactors for rapid and efficient sample preparation in MS-based proteomic studies. Proteomics 13(3–4), 457–466 (2013).
- Spross J , Sinz A. A capillary monolithic trypsin reactor for efficient protein digestion in online and offline coupling to ESI and MALDI mass spectrometry. Anal. Chem. 82(4), 1434–1443 (2010).
- Kim JH, Inerowicz HD, Hedrick V, Regnier FE. Integrated sample preparation methodology for proteomics: analysis of native proteins. Anal. Chem. 85(17), 8039–8045 (2013).
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 6(5), 359–362 (2009).
- Maurer M, Muller AC, Wagner C et al. Combining Filter-aided sample preparation and pseudoshotgun technology to profile the proteome of a low number of early passage human melanoma cells. J. Proteome Res. 12(2), 1040–1048 (2013).
- Magni F, van der Burgt YE, Chinello C et al. Biomarkers discovery by peptide and protein profiling in biological fluids based on functionalized magnetic beads purification and mass spectrometry. Blood Transfus. 8 Suppl 3, s92–s97 (2010).
- Peter JF , Otto AM. Magnetic particles as powerful purification tool for high sensitive mass spectrometric screening procedures. Proteomics 10(4), 628–633 (2010).
- Bladergroen MR, Derks RJ, Nicolardi S et al. Standardized and automated solid-phase extraction procedures for high-throughput proteomics of body fluids. J. Proteomics 77, 144–153 (2012).
- Luckwell J, Beal A. Automated micropipette tip-based SPE in quantitative bioanalysis. Bioanalysis 3(11), 1227–1239 (2011).
- Callesen AK, Madsen JS, Vach W, Kruse TA, Mogensen O, Jensen ON. Serum protein profiling by solid-phase extraction and mass spectrometry: A future diagnostics tool? Proteomics 9(6), 1428–1441 (2009).
- Mommen GP, Meiring HD, Heck AJ, de Jong AP. Mixed-bed ion exchange chromatography employing a salt-free pH gradient for improved sensitivity and compatibility in MudPIT. Anal. Chem. 85(14), 6608–6616 (2013).
- Motoyama A, Xu T, Ruse CI, Wohlschlegel JA, Yates JR, III. Anion and cation mixed-bed ion exchange for enhanced multidimensional separations of peptides and phosphopeptides. Anal. Chem. 79(10), 3623–3634 (2007).
- Welsch N, Lu Y, Dzubiella J, Ballauff M. Adsorption of proteins to functional polymeric nanoparticles. Polymer 54, 2835–2849 (2013).
- Longo C, Patanarut A, George T et al. Core-shell hydrogel particles harvest, concentrate and preserve labile low abundance biomarkers. PLoS One. 4(3), e4763 (2009).
- Clonis YD. Affinity chromatography matures as bioinformatic and combinatorial tools develop. J. Chromatogr. A 1101(1–2), 1–24 (2006).
- Polaskova V, Kapur A, Khan A, Molloy MP, Baker MS. High-abundance protein depletion: comparison of methods for human plasma biomarker discovery. Electrophoresis 31(3), 471–482 (2010).
- Ogata Y, Charlesworth MC, Muddiman DC. Evaluation of protein depletion methods for the analysis of total-, phospho- and glycoproteins in lumbar cerebrospinal fluid. J. Proteome Res. 4(3), 837–845 (2005).
- Qian WJ, Kaleta DT, Petritis BO et al. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol. Cell Proteomics 7(10), 1963–1973 (2008).
- Halfinger B, Sarg B, Amann A, Hammerer-Lercher A, Lindner HH. Unmasking low-abundance peptides from human blood plasma and serum samples by a simple and robust two-step precipitation/immunoaffinity enrichment method. Electrophoresis 32(13), 1706–1714 (2011).
- Hao P, Guo T, Sze SK. Simultaneous analysis of proteome, phospho- and glycoproteome of rat kidney tissue with electrostatic repulsion hydrophilic interaction chromatography. PLoS One 6(2), e16884 (2011).
- Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J. Proteome Res. 3(2), 235–244 (2004).
- Ackermann BL , Berna MJ. Coupling immunoaffinity techniques with MS for quantitative analysis of low-abundance protein biomarkers. Expert Rev. Proteomics 4(2), 175–186 (2007).
- Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An Automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics 9(1), 184–196 (2010).
- Wilson R. Sensitivity and specificity: twin goals of proteomics assays. Can they be combined? Expert Rev. Proteomics 10(2), 135–149 (2013).
- Selman MHJ, Hemayatkar M, Deelder AM, Wuhrer M. Cotton HILIC SPE microtips for microscale purification and enrichment of glycans and glycopeptides. Anal. Chem. 83(7), 2492–2499 (2011).
Information resources
Table 1: Various sorbents used in protein solid-phase extraction (SPE).
| SPE type | Material | Usage |
| Reversed phase | C3, C4, C8 | Proteins |
| C18 | Peptides | |
| Normal phase | ZIC-HILIC | Glycoproteins, glycopeptides |
| TSKgel Amide-80 | Proteins | |
| Waters HILIC | Proteins, peptides | |
| Cotton* | Glycopeptides, glycans after release from peptides | |
| IEX | WCX, SCX | Proteins |
| WAX, SAX | Proteins | |
| Metal-chelating | Ti, Fe | Phosphopeptides |
| Ga | Phosphopeptides | |
| Cu | (Phospho)peptides | |
| Affinity | Lectin | Glycans, glycopeptides |
| Boronic acid | Glycans, glycopeptides | |
| Blue dye | Albumin | |
| Protein A/G | Immunoglobulins | |
| Heparin | IgG | |
| RNA/DNA | Plasmids, DNA binding proteins | |
| Purine/pyrimidine derivatives | e.g., ATP/GTP using enzymes | |
| Coenzymes | Coenzyme-dependent enzymes | |
| Vitamins | Vitamin binding proteins | |
| Antibodies | Proteins, peptides |
For further information, see [29] and references cited therein.
* Reference [44].
Box 1: Factors to consider for sample collection.
