Chapter 5: Modern techniques in quantitative proteomics
Doi: 10.4155/FSEB2013.13.124
Authors
Jeffrey Charles Smith
Department of Chemistry
Carleton University
1125 Colonel By Drive
Ottawa, ON K1S 5B6
Tel: 1-613-520-2600 x2408
Fax: 1-613-520-3749
E-mail: [email protected]
Andrew MacKendrick Macklin
Department of Chemistry
Carleton University
1125 Colonel By Drive
Ottawa, ON K1S 5B6
Tel: 1-613-520-2600 x2408
Fax: 1-613-520-3749
E-mail: [email protected]
About the Authors
Jeffrey C. Smith
Jeff Smith is an Associate Professor in the Department of Chemistry and Institute of Biochemistry and the Director of the Carleton Mass Spectrometry Centre at Carleton University in Ottawa, Ontario, Canada. His research program includes the use of mass spectrometry and microfluidics to develop novel quantitative proteomic and lipidomic strategies, and applying these to monitor the dynamics of proteins and lipids from a systems biology point of view. A relatively new academic, Jeff has published 36 articles that have collectively been cited over 1000 times on the use of mass spectrometry-based proteomics and lipidomics.
Andrew M. Macklin
Andrew completed his MSc in chemistry at Carleton University under the supervision of co-author Dr. Jeff Smith. His research involved characterizing organic molecules that enhance viral replication for cancer therapeutic and vaccine production applications by means of mass spectrometry and proteomic methods. Currently, Andrew is a research assistant at York University where he manages an Orbitrap mass spectrometer facility.
Modern techniques in quantitative proteomics
Introduction to quantitative Proteomics
The field of proteomics has expanded at an impressive rate over the past 15 years as traditional quantitative methods such as ELISA and western blotting are being replaced by a new golden standard: the mass spectrometer. The rise of mass spectrometry (MS) in the laboratory has coincided with a significant shift from qualitative to quantitative MS-based proteomics in the literature. Rather than providing a list of proteins identified in a sample, quantitative proteomics has been fundamental in enhancing our understanding of protein expression and how their abundances can fluctuate. A typical study compares protein expression under homeostatic conditions to the abundance of the same proteins in response to an extracellular stimulus.
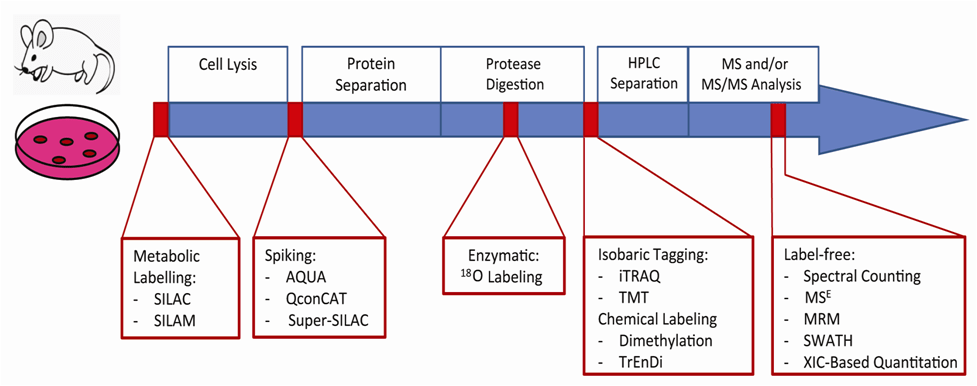
Described in the following sections is a selection of peptide quantitation techniques that are currently being used and, in many cases, continuously improved (Figure 1). Many of these methods involve labeling proteins with heavy and light stable-isotope pairs. Proteins can be metabolically labeled with heavy or light isotope-containing growth media or derivatization can occur following proteolytic digestion with the use of isotopically distinct chemical labels or isobaric tags. Other quantitation techniques eliminate labeling (label-free) and rely on advanced software analysis. These methods measure the relative concentrations of peptide analytes within two or more samples. In contrast, absolute quantitation techniques make use of internal standard peptides that have been synthetically prepared for selected or multiple reaction monitoring (SRM or MRM, respectively) analysis and are quickly gaining popularity. The reader should realize from this chapter that there are a variety of innovative quantitative proteomic methods available and that the advantages and disadvantages of each must be taken into consideration for proper application. As more experimental strategies build upon a solid understanding of this field, MS-based peptide quantitation will continue to yield fascinating results for years to come as it is used to study the dynamics of living systems.
Figure 1: The incorporation of quantitation strategies into a typical proteomics workflow. A quantitative proteomics workflow consists of cellular lysis, protein separation and digestion followed by LC-MS analysis. There are multiple time-points at which peptide quantitation strategies may be introduced. Initial cell culture or animal model samples can be labeled metabolically at the protein level. Following lysis, labeled proteins may be spiked in or enzymatic labeling may occur during digestion. Further downstream, peptides can be labeled chemically or isobarically. Label-free techniques require the least sample preparation; quantitation occurs during or after data analysis.
Metabolic-based labeling
For the majority of relative quantitation techniques, the incorporation of unique isotopes occurs after cell lysis as the sample is being prepared for MS analysis. By comparison, metabolic labeling strategies incorporate isotopic labels into all proteins during cellular growth. Stable-isotope labeling with amino acids in cell culture (SILAC) makes use of growth medium containing isotopically distinct amino acids, which are incorporated into proteins within a few generations of cell division [1]. Isotopically labeled arginine and lysine are common vehicles for incorporating the label considering that typical proteomic workflows involve tryptic digestion, yielding peptides that terminate with one of these two amino acids.
SILAC’s utility in proteomics has been demonstrated by numerous exemplary studies. One advantage of this quantitative approach is the elimination of indeterminate error that arises from sample handling, since the differentially labeled samples are prepared identically and combined following cell lysis. This makes SILAC a favorable technique to study post-translational modifications as these studies often require substantial sample handling (e.g., affinity enrichment steps) following cell lysis [2].
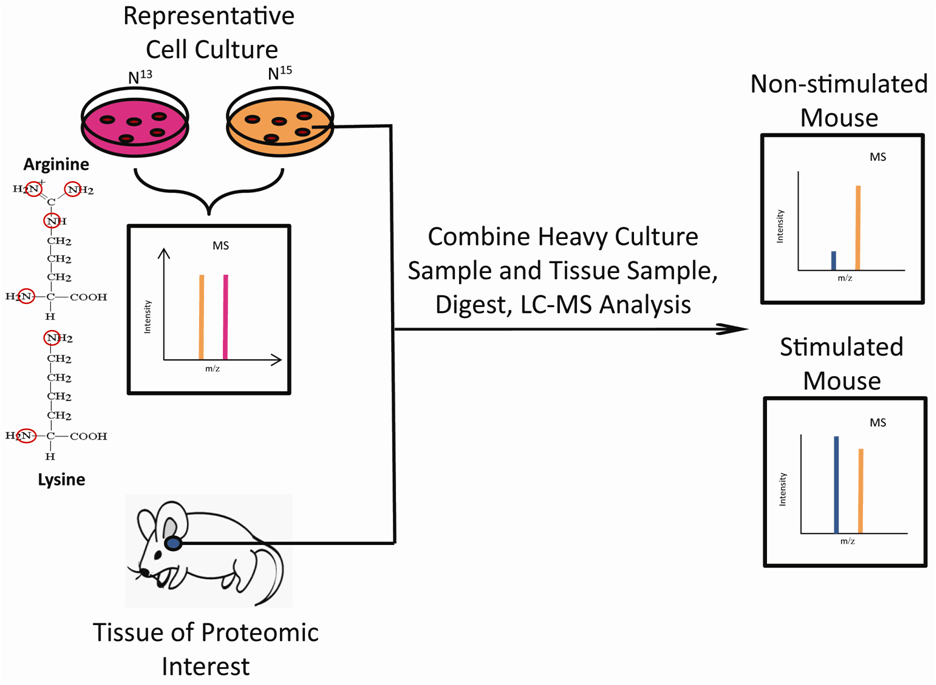
Initially, SILAC was limited to experiments involving cell culture; however, recent developments have extended its use to in vivo studies. The Super-SILAC method produces isotopically labeled cell lysates that can be mixed with non-labelled tissue lysates to enable relative quantitation (Figure 2). Monetti et al. made use of this strategy to quantify 10,000 phosphorylation sites associated with the insulin response in mouse hepatocytes. Their efforts resulted in the creation of an accurate quantitative phosphoproteomic dataset for future reference [3]. SILAC-based strategies are also being developed to impact research using human embryonic stem cells (hESC). hESC culturing systems employ a feeder layer of cells that release non-labeled amino acids into the media thereby complicating the SILAC process. Liberski et al. created a feeder-layer free system and unique growth media to make the culturing of hESCs amenable to the SILAC process [4]. This technology presents the opportunity for SILAC to investigate the valuable mechanisms underlying hESC self-renewal and differentiation.
Despite being primarily used in cell culture, metabolic labeling is also applicable to model organisms, such as rodents, plants and insects. Using deuterated feed, mice have been metabolically labeled to demonstrate that protein turnover rates differ among tissues [5]; the rate in muscle was slower than liver, which was followed by the heart. This method, termed stable isotope labeling in mammals (SILAM), is still in its infancy owing to high monetary and temporal expenses. Furthermore, the number of 15N labels varies from peptide to peptide. The MS data produced by this method is hence more complicated and demands rigorous data analysis schemes, which make 15N-labelling less appealing. Regardless, metabolic labeling is providing researchers with newly developed strategies at an accelerating rate to quantify proteomic dynamics.
Figure 2: Super-SILAC metabolic labeling strategy. SILAC quantitation is typically limited to cell culture using light (14N) or heavy (15N) versions of arginine and lysine. Peptides from light and heavy cultures are combined to create two distinct MS peaks revealing relative intensities. The Super-SILAC method extends this technique to extracted tissue samples. The tissue of interest is mixed with cells that were cultured in heavy media. The heavy cellular lysate (orange peaks) and tissue lysate (blue peaks) are combined, digested and the relative quantities of identical peptides from each source are revealed by MS.
Isobaric mass tagging
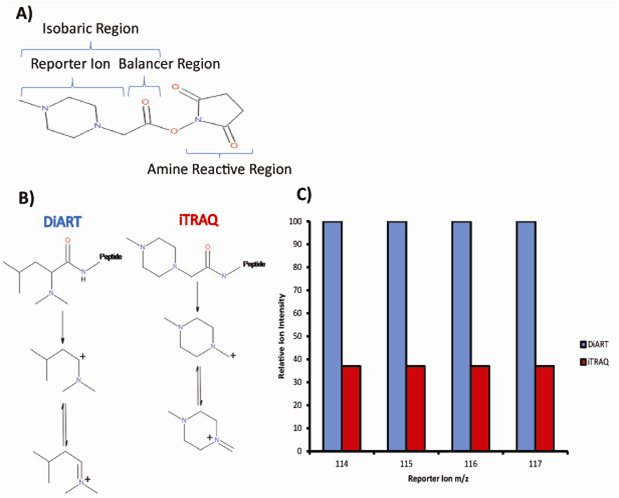
A relative quantitation technique that is becoming more prominent in the proteomic literature is isobaric tagging. In contrast to metabolic labeling, the tagging step takes place following protein digestion. The two most prevalent isobaric tag technologies include isobaric tags for relative and absolute quantitation (iTRAQ) and tandem mass tags (TMT). The general structures for these tags include a reporter region, a balancing region and an amine-reactive region (Figure 3A). One key difference from metabolic labeling is that the relative quantitation is observed in the MS2 spectra since the labeled peptides fragment to conveniently produce ions representing the isotopically labeled reporter region [6].
iTRAQ technology is not restricted to specific sample types, allowing for numerous areas of application. The value of iTRAQ has been demonstrated many times in vitro and has begun to emerge in clinical analyses. For example, current methods to detect the tumor-inducing virus human papillomavirus (HPV), including hybridization and real-time polymerase chain reaction (RT-PCR) have been deemed relatively inadequate compared to MS-based iTRAQ methods [7]. In a recent example, 8-plex iTRAQ was used to label tryptically digested cervical smears for analysis via MS; the in-depth proteome coverage allowed for the confident relative quantitation of six different HPV proteins [7]. Similar examples illustrate how quantitative proteomic methods are improving the quality and breadth of diagnostic procedures in the clinical laboratory.
Isobaric mass tagging has also been used to improve industrial processes, such as the production of ethanol. Cyanobacterial species that are used to produce biofuels are negatively impacted by high concentrations of the ethanol they secrete, thus restricting production. Engineering new strains of biofuel-producing cyanobacteria represents significant value from environmental and economical points of view; however, the underlying protein mechanisms governing the cellular response to environmental ethanol have been poorly understood. In an effort to identify ethanol tolerance mechanisms and possible targets for genetic manipulation, the iTRAQ 8-plex kit was used to investigate the cyanobacterial response to ethanol [8]. The study identified 42% of the cyanobacterial proteome and revealed that the expression of 293 proteins changed in a significant manner, highlighting potential gene targets for further investigation and industrial optimization.
Another novel application of isobaric tagging is the identification of biomarkers so that disease progression may be monitored. Tsuchida et al. discovered that neutrophil protein LCN-2 became more abundant as periodontal disease progressed. Experiments were conducted using TMT to label and compare proteins in gingival fluid from both healthy and diseased patients. With supporting evidence from traditional proteomic methods, it was the first comprehensive view of the relative abundance of gingival fluid proteins in relation to periodontal disease [9]. Pichler et al. compared the effectiveness of the TMT and iTRAQ tagging kits in terms of peptide identification and found that the 4-plex iTRAQ tag led to the identification of approximately 1.7-times more peptides than the 6-plex TMT tag [10].
Despite its relatively low cost, many quantitative proteomics strategies avoid the use of deuterium as a heavy isotope owing to the deuterium effect; deuterium and hydrogen exhibit different adsorbances in reversed-phase chromatography. This is especially undesirable for isobaric tagging, which relies on the mixing of differentially labeled peptide samples, thus co-elution of identical peptides is essential for accurate quantitation. It was found that the deuterium effect is inconsequential when the labels are in close proximity to hydrophilic groups. This finding opened the door for a new isobaric mass tag: the deuterium isobaric amine reactive tag (DiART). Peptide-bound DiART fragmentation yields a secondary carbocation that is more stable than the primary carbocation produced by iTRAQ. This causes more frequent fragmentation of the bond that releases the reporter ion thus allowing for improved sensitivity (Figure 3B). Chen et al. compared the application of iTRAQ and DiART to the quantitation of thermophilic bacterial proteins and concluded that DiART showed improved reporter ion intensity, therefore enabling more accurate relative quantitation (Figure 3C) [11].
Isobaric mass tagging continues to be an innovative method for relative quantitation in proteomics. Experiments rely on collision induced dissociation (CID), which produces spectra that simultaneously provide quantitative and identification information for each peptide in an experiment. Combining CID with higher-energy collisional dissociation (HCD) on Orbitrap instruments is gaining popularity as it significantly improves the accuracy of peptide quantitation. This comes with the consequence of longer scan times that limit the number of peptides detected [12]. Continual improvements in reagent costs and protocol reproducibility will lead to an increased prevalence of this technique in both the literature and in the clinic.
Figure 3: iTRAQ (isobaric tags for relative and absolute quantitation) structure and comparison to DiART (deuterium isobaric amine reactive tag). A) The iTRAQ tag’s structure is composed of an isobaric region and an amine reactive region responsible for covalent modification of peptide N-termini. The isobaric region consists of a reporter region that produces fragment ions with mass to charge ratios of 114–117 Th and a complementary balancing region. B) DiART is another isobaric tag that makes use of deuterium labels as opposed to 13C or 15N. The production of reporter ions in iTRAQ and DiART is illustrated. C) On average, the intensities of the reporter fragment ions produced via DiART are 2.7× higher than iTRAQ. Reproduced from [11] with permission. © ACS (2012).
Chemical and enzymatic derivatization
Similar to labeling with isobaric tags, chemical derivatization occurs at the peptide level. The main difference from isobaric tagging is that quantitation is achieved using the MS spectra, rather than the MS2 spectra. Chemical derivatization of peptides involves adding a label containing a stable isotope in a predictable manner. The most prevalent derivatization strategy in the literature is dimethyl labeling, a technique that is faster and significantly cheaper than isobaric tagging. In the presence of cyanoborohydride, this strategy uses 12C/13C-containing or hydrogen/deuterium-containing formaldehyde to reductively dimethylate primary amines (peptide N-termini and lysine side chains). Mixing light- and heavy-labeled samples reveals a mass discrimination of 4 and 8 Da for arginine- and lysine-terminating peptides, respectively. This method is particularly well suited to applications that are not feasible with metabolic labeling, such as in the case of human tissue samples [13].
Mesenchymal stem cells (MSCs) show therapeutic potential for repairing tissues and treating diseases; however, historically there has been difficulty identifying the protein pathways responsible for the pluripotency of these cells. In a study by She et al., dimethyl labeling was used to compare the relative abundances of thirty-four MSC proteins to protein isoforms found in differentiated cells to elucidate differentiation pathways. The technique was effective despite the high sequence homology and large concentration range of protein analytes in their study [14].
Dimethyl labeling has also been used to study the post-translational modification (PTM) ubiquitination. Tryptic digestion of ubiquitinated proteins yields isopeptides that include a trademark diglycine branch. Despite the uniqueness of this occurrence, false-positive results are frequently observed. It was proposed that the additional N-terminus possessed by these distinctive peptides can be used for additional methylations to enhance their detection in complex samples [15]. Dimethyl labeling also enhances a1 and b2 signal peaks upon CID, which allowed for the improved sequencing of diglycine-branched isopeptides.
Chemical derivatization is often multiplexed with other labeling strategies, such as SILAC. As shown by Wang et al., this powerful combination of stable-isotope labeling strategies permitted the quantitation of six samples, as opposed to a maximum of three for each technique. To further the experiment, this procedure was used to accurately determine the 50% turnover rate of more than 1300 proteins [16].
Stable-isotope labels may also be incorporated into peptides enzymatically through the use of trypsin and 18O-labeled water. The proteolytic mechanism of trypsin includes the transfer of two oxygen atoms to the C-terminus of the generated peptides. Therefore, relative quantitation can be performed by comparing the ratios of the 16O/18O labeled peptide pairs that are distinguishable by 4 Th in the MS spectra. This method was recently used to identify proteins associated with biofilm formation and gingivitis [17]. Through a comparison of the infectious and non-infectious strains of gingivitis-causing bacteria, a family of C-terminal domain proteins was identified to increase upon biofilm formation thus providing an opportunity to target these proteins.
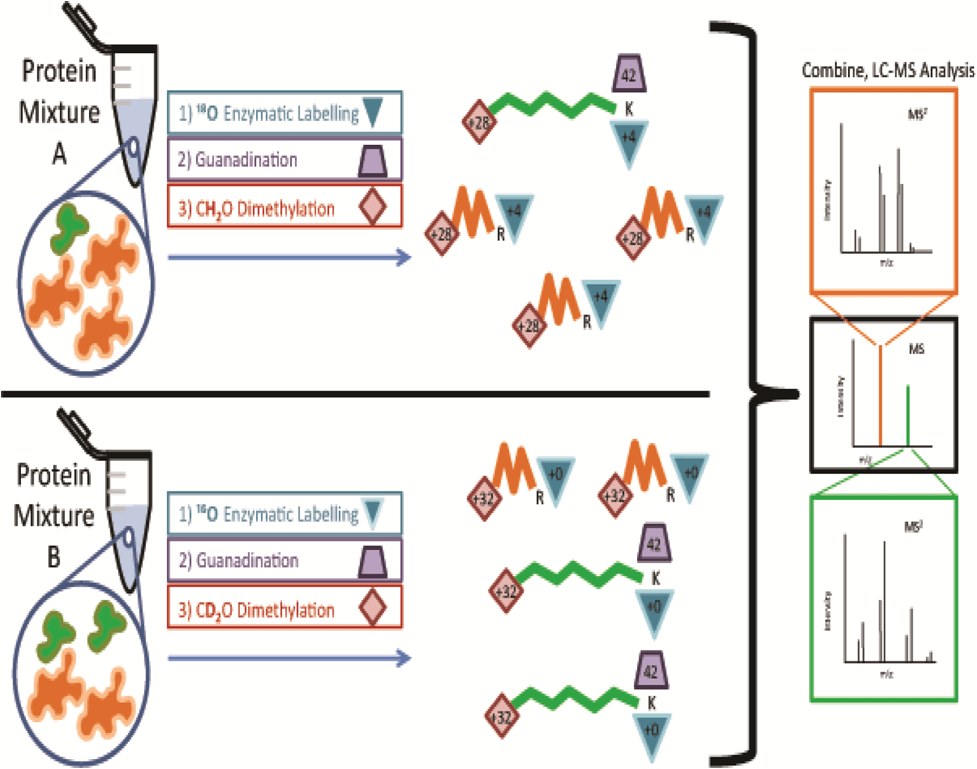
An issue with chemical or enzymatic quantitation methods is that differentially labeled peptides with identical sequences appear as distinct peaks in each spectrum requiring additional analysis time. Yang et al. developed a solution, termed quantitation by isobaric terminal labeling (QIRT), which multiplexes enzymatic labeling with reductive methylation to produce isobaric peptides (Figure 4). Hepatocellular carcinoma proteins were tryptically digested in 16O/18O water and the lysine residues guanidinated to prevent further reaction. Following this, peptides were labeled with heavy or light versions of formaldehyde to counteract the additional mass introduced by the oxygen label. This produced isobaric peptides that could be accurately quantified by their MS2 spectra with increased sensitivity [18].
Enzymatic labeling has not become as prominent in the literature owing to the small mass shift and gradual back-exchange of 18O/16O. To overcome these challenges, Pan et al. dissolved trypsin in an organic solution and used its ligase activity; rather than have it act as a protease. Peptides that had been previously digested by trypsin in aqueous solution could be modified with the addition of a heavy-isotope labeled amino acid [19]. This innovative new method could be coupled with SILAC to allow greater multiplexing and increase the prominence of enzymatic labeling.
Figure 4: QIRT (quantitation by isobaric terminal labeling) labeling strategy. Two protein samples are isobarically tagged by combining enzymatic and chemical labeling techniques. One sample is tryptically digested in the presence of 18O-labelled water (+4 Th), while the other is digested in unlabeled water (+0 Th). Both samples are subjected to guanidination (+42 Th) to protect amine groups on lysine (K) side chains that would otherwise be reactive with subsequent dimethyl labeling. Arginine (R) side chains remain unaffected. Dimethyl labeling is used to incorporate heavy (+32 Th) and light (28 Th) versions of formaldehyde into the C-termini of peptides to counter-balance additional mass from enzymatic labeling. Once the peptide samples are combined, peptides of identical sequence are represented by one MS peak. Peptide identification and accurate relative quantitation is permitted by comparing intensities of y and b ion pairs separated by m/z 4 in the MS2 spectra. In this example, orange peptides are more abundant in Mixture A and green peptides are more abundant in Mixture B. These relative abundances are reflected in the MS2 spectra where heavier peak in each fragment ion pair is representative of the peptide that originated from Mixture B. Reproduced from [18] with permission. © Elsevier (2012).
Label-free quantitation
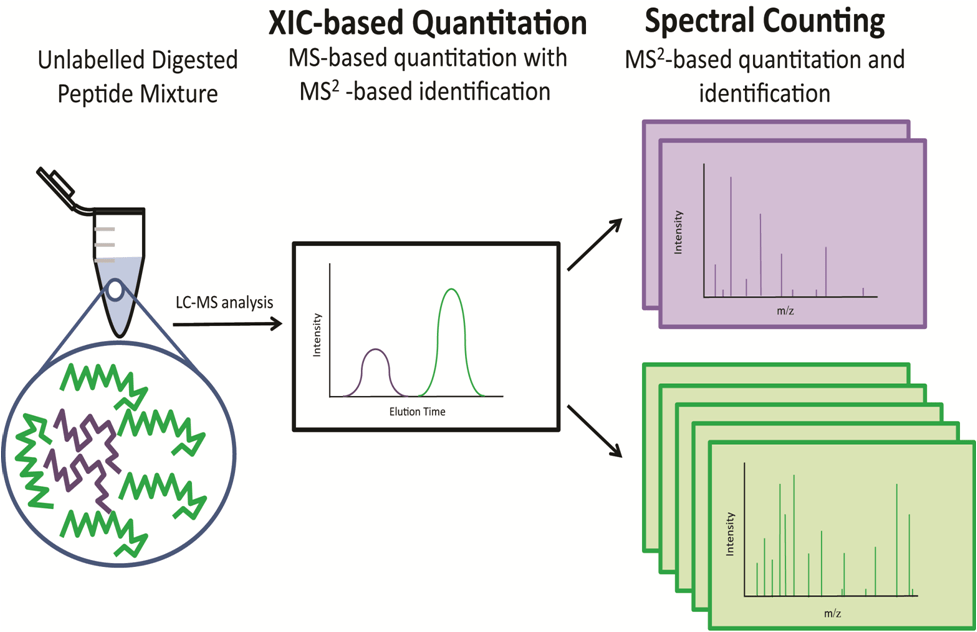
Label-free quantitation is faster and cheaper than isotopic labeling strategies and offers exceptional suitability for high-throughput global proteome analysis. There are two major categories of label-free methods: extracted ion chromatogram (XIC)-based quantitation, and spectral counting (Figure 5).
XIC-based quantitation is based on the assumption that higher peptide concentrations will generate a greater area underneath the integrated chromatographic peak from MS spectra. Meanwhile, peptide identification is still possible through MS2 spectra. Computational methods are greatly improving the “clean-up” of data concerning this method including elution peak alignment, peak normalization, peak picking and noise suppression [20]. XIC-based quantitation in proteomics experiments often relies on MRM-based methods for improved selectivity and attomolar sensitivity [20]. These methods use several parent-to-fragment ion transitions for each peptide to avoid isobaric interference allowing many proteins to be monitored throughout the course of an experiment. MRM experiments can be coupled to data-dependent acquisition (DDA) schemes whereby MRM signal triggers acquisition of an MS2 spectrum on the parent mass [20]. Owing to the enhanced sensitivity of MRM, label-free quantitation is often used as a tool to validate preliminary quantitative proteomics experiments. MRM label-free quantitation methods demonstrate greater sensitivity, reproducibility and dynamic range than non-targeted MS methods; however, they have historically lacked the ability to quantify a large set of proteins.
One challenge with combining XIC-based quantitation with DDA-based identification is finding a balance between MS (quantitation) and MS2 (identification) cycling. Devoting more scanning time to peptide identification decreases the number of data points available for MS quantitation, which limits the resolution, and hence accuracy of XIC-based quantitation. MSE was developed as a data-independent acquisition (DIA) method to overcome this hurdle. In MSE, all peptides are fragmented regardless of abundance as the mass spectrometer cycles between high and low collision energies [20]. Peptides are identified by aligning precursor and fragment ion chromatographic elution profiles and quantitation is achieved by determining the area under the peak for the precursor ions XICs.
Another unique DIA method that has been recently developed expands the precision of MRM to large complex peptide samples termed MSALL or SWATH-MS [21]. Historically, conducting MS2 on several precursor ions at the same time has resulted in complex spectra that could not be interpreted. SWATH-MS, however, rapidly cycles through 25 Th windows within a set range and fragments all precursor ions. Again, mapping the chromatographic elution profiles of each fragment and parent ion elucidates which fragments arose from each parent ion for identification; the data may then be mined post hoc to create MRM transitions for precise quantitation as well as precursor or neutral loss spectra. SWATH-MS was used to detect a higher number of N-linked glycoproteins than MRM methods without significantly sacrificing sensitivity and reproducibility [22].
One area of research where this technology is being applied is the study of genetically modified crops (GMCs). A growing global demand for food places greater emphasis on the development and improvement of GMCs. Consequentially, this creates a demand for accurate analyses that can verify that unintentional changes are not made to crops after genetic manipulation. An in silico digest of the soybean proteome provided a list of expected peptides and MSE successfully detected the majority of the soybean proteome with high sequence coverage [23].
Wasslen et al. recently reported a technique termed TrEnDi that uses diazomethane to enhance label-free quantitation via modifying peptides such that they obtain a fixed, permanent positive charge [24]. The modified peptides exhibit reduced ion suppression since proton affinity is not critical for ionization and they dissociate to form intense a2 fragments. TrEnDi increases ionization efficiency and leads to more sensitive as well as fully predictable MRM transitions, enhancing and simplifying label-free quantitative proteomics workflows [25].
Spectral counting is based on the expectation that the number of peptide-identifying MS/MS spectra correlates directly with the abundance of protein. It utilizes normalization factors, such as exponentially modified protein abundance index (emPAI), absolute protein expression (APEX) and normalized spectral abundance factor (NSAF), which account for the physicochemical properties and lengths of peptides with respect to their abilities to be ionized [26]. For example, normalized spectral index (SIN) was developed to greatly improve the reproducibility of label-free quantitation by considering spectral counting in conjunction with peptide count and fragment ion intensity [27]. With the ongoing improvement of data analysis software, label-free quantitation will still hold advantages over label-dependent approaches and continue to be a reliable way to monitor protein dynamics in a given system. Label-free strategies are more commonly used for relative quantitation but can also be employed for absolute quantitation through the addition of internal standards with known response factors or via the creation of a standard curve
Figure 5: Common label-free quantitation strategies using standard addition methods. An unlabeled peptide mixture containing green and purple peptides can be relatively quantified via extracted ion chromatogram (XIC)-based quantitation or spectral counting. In this diagram, the green peptide is 2.5× more abundant than the purple peptide and this relative abundance is reflected by both the XIC peak area and the number of MS2 spectra that successfully identify the peptide.
Absolute quantitation methods
Relative quantitation techniques compare the intensities of differentially labeled peptides in the context of complex samples. Since the concentration of peptides arising from separate complex samples is unknown, actual peptide concentrations cannot be concluded by comparing the intensities of two peaks. Absolute quantitation techniques, however, determine peptide concentrations by spiking the sample of interest with known concentrations of one or more heavy isotopically labeled standard peptides. Spiked peptides are synthetically prepared according to the absolute quantitation (AQUA) method. Alternatively the quantification concatamer (QconCAT) method incorporates bacteria grown in SILAC medium that can be transformed with high expression vectors to produce artificial proteins composed of standard peptides. Absolute quantitation methods often utilize targeted MRM methods for optimal sensitivity to determine the concentrations of low abundance peptides.
Methods currently used to quantify peptides presented by major histocompatbility complex (MHC) molecules on the surface of leukocytes rely on monoclonal T-cell stocks and antibodies, which require expensive and laborious cell culture practices. AQUA was recently demonstrated to be a higher-throughput and less expensive alternative for quantifying peptides antigens [28]. Once the most intense transitions for the peptide of interest were chosen, the method enabled the quantitation of over one hundred peptide epitopes. The authors believe that AQUA-MRM has the ability to revolutionize many immunological assays that require laborious upkeep of monoclonal cell lines and lack absolute quantitation.
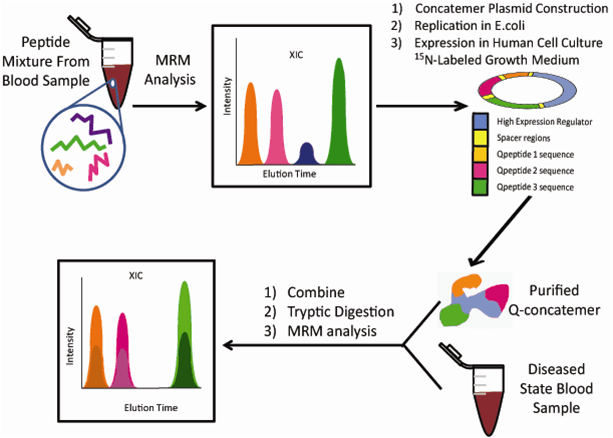
The QconCAT method is more economical for studies that investigate the absolute abundances of a high number of proteins (Figure 6). This methodology was very useful in evaluating the stoichiometry of the glycolysis pathway in yeast [29]. A large concatemer was engineered to generate at least three peptides from 27 different isoenzymes involved in the pathway. All enzymes were successfully quantified within a dynamic range of 14,000 and ten million molecules per cell, therefore providing accurate information for future bioinformatic in silico modelling. Furthermore, the absolute quantitation data was compared to label-free data and it was observed that the label-free approach underestimates the abundances of proteins.
One drawback to these quantitative workflows is the lengthy optimization process, which restricts the throughput of method development. This involves identifying a potential Q-peptide and optimizing the MS-parameters for improved sensitivity. As these absolute quantitation methods gain popularity, more computer algorithms are being developed to facilitate this limiting step. CONseQENCE is an example of a Q-peptide predictor that accounts for peptide characteristics such as hydrophobicity, charge, size and secondary structure [30]. With computational method development, the optimization process will become simplified and pave the way for absolute quantitation to become more accessible in the future.
Figure 6: The quantification concatamer (QconCAT) strategy. MRM-based methods are first optimized for peptides from digested blood. To permit absolute quantitation, a large synthetic protein (Q-concatemer) is constructed, transfected into Escherichia coli and then translated to human cells growing in 15N-labeled growth media. The purified Q-concatemer is spiked into sample blood at a known concentration, which is then digested permitting MRM-based absolute quantitation of blood proteins/peptides.
Conclusion
There are many recent strategies to quantify protein samples using a mass spectrometer. Improvements in sensitivity, multiplexing, sample manipulation, reagent cost and time requirements are continuously being observed. The field has recently witnessed a trend towards the simultaneous use of more than one quantitative method to combine the benefits of each technique. There has also been a tendency towards absolute quantitation as the cost of reagents decrease and as new techniques in this area emerge. The growing prominence of literature in the field of quantitative proteomics over the past decade demonstrates the value of research in this area and implicates the importance of this field in understanding the dynamics of proteins from a biological systems perspective.
Executive summary
- Mass spectrometry-based proteomics is undergoing a transition from qualitative to quantitative approaches to further our knowledge of biological systems. This will benefit many areas of research including the discovery of biomarkers, the optimization of industrial processes, the use of mass spectrometry-based technology in clinical diagnosis, the development of pharmaceutical sciences, and investigations into food science and crop engineering.
- Metabolic labeling incorporates distinct isotopes into proteins by culturing cells in isotopically defined media, which ultimately reduces the experimental indeterminate error compared to other quantitative proteomics strategies.
- Chemical and enzymatic derivatization of peptides, such as dimethyl labeling, are quick and inexpensive alternatives that can be combined with other labeling techniques for expanded multiplexing.
- Isobaric tagging is a powerful technique for sensitive relative quantitation between samples. This technique relies on the fragmentation of a covalently bound label that produces reporter ions observable in MS2
- Label-free quantitation encompasses data-independent acquisition (DIA) and data-dependent acquisition (DDA) strategies that entail less sample preparation but involve more demanding data analysis schemes.
- Absolute quantitation techniques accurately determine protein concentrations but require intensive method optimization prior to sample analysis.
References
- Ong S-E. The expanding field of SILAC. Bioanal. Chem. 404, 967–976 (2012).
- Stoehr G, Schaab C, Graumann J, Mann M . A SILAC-based approach identifies substrates of caspase dependent cleavage upon TRAIL-induced apoptosis. Cell. Proteomics 12 (6), 1–35 (2013).
- Monetti M, Nagaraj N, Sharma K, Mann M. Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Methods 8 (8), 655–658 (2011).
- Liberski AR, Al-Noubi MN, Rahman ZH et al. Adaptation of a commonly used, chemically defined medium for human embryonic stem cells to stable isotope labeling with amino acids in cell culture. J. Proteome Res. 12 (7), 3233–3245 (2013).
- Claydon AJ, Thom MD, Hurst JL, Beynon RJ. Protein turnover: Measurement of proteome dynamics by whole animal metabolic labeling with stable isotope labeled amino acids. Proteomics 12, 1194–1206 (2012).
- Evans C, Noirel J, Ow SY et al. An insight into iTRAQ: where do we stand now? Bioanal. Chem. 404, 1011–1027 (2012).
- Papachristou EK, Roumeliotis TI, Chrysagi A et al. The shotgun proteomic study of the human ThinPrep cervical smear using iTRAQ mass-tagging and 2D LC-FT-Orbitrap-MS: the detection of the human papillomavirus at the protein level. Proteome Res. 12, 2078–2089 (2013).
- Qiao J, Wang J, Chen L et al. Quantitative iTRAQ LC−MS/MS proteomics reveals metabolic responses to biofuel ethanol in cyanobacterial synechocystis sp. PCC 6803. Proteome Res. 1, 5286–2300 (2012).
- Tsuchida S, Satoh M, Kawashima Y et al. Application of quantitative proteomic analysis using tandem mass tags for discovery and identification of novel biomarkers in periodontal disease. Proteomics 0, 1–12 (2013).
- Pichler P, Kocher T, Holtzmann J et al. Peptide labelling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Chem. 82, 6549–6558 (2010).
- Chen Z, Wang Q, Lin L et al. Comparative evaluation of two isobaric labeling tags, DiART and iTRAQ. Chem. 84, 2908–2915 (2012).
- Kocher T, Pichler P, Schutzbier M et al. High precision quantitative proteomics using iTRAQ on an LTQ Orbitrap: A new mass spectrometric method combining the benefits of all. J. Proteome Res. (2009).
- Kovanich D, Cappadona S, Raijmakers R, Mohammed S, Scholten A, Heck AJ. Applications of stable isotope dimethyl labeling in quantitative proteomics. Bioanal. Chem. 404, 991–1009 (2012)
- She Y-M, Rosu-Myles M, Walrond L, Cyr TD. Quantification of protein isoforms in mesenchymal stem cells by reductive dimethylation of lysines in intact proteins. Proteomics 12, 369–379 (2012).
- Chicoree N, Connolly Y, Tan C-T et al. Enhanced detection of ubiquitin isopeptides using reductive methylation. Am. Soc. Mass Spectrom. 24, 421–430 (2013).
- Wang F, Cheng K, Wei X et al. A six-plex proteome quantification strategy reveals the dynamics of protein turnover. Rep. 3, 1827–1833 (2013).
- Ang C-S, Veith PD, Dashper SG, Reynolds EC. Application of 16O/18O reverse proteolytic labeling to determine the effect of biofilm culture on the cell envelope proteome of Porphyromonas gingivalis W50. Proteomics 8, 1645–1660 (2008).
- Yang S-J, Nie A-Y, Zhang L et al. A novel quantitative proteomics workflow by isobaric terminal labeling. Proteomics 75, 5797–5806 (2012).
- Pan Y, Ye L, Xhao L et al. N-terminal labeling of peptides by trypsin-catalyzed ligation for quantitative proteomics. Angew. Chem. Int. Ed. 2, 1–6 (2013).
- Neilson KA, Ali NA, Muralidharan S et al. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 (2011).
- Liu Y, Huttenhain R, Surinova S. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 13(8). 1247–1256 (2013).
- Megger DA, Bracht T, Meyer HE et al. Label-free quantitation in clinical laboratories. Biophys. Acta 1834, 1581–1590 (2013).
- Murad AM, Rech EL. NanoUPLC-MSE proteomic data assessment of soybean seeds using the Uniprot database. BMC Biotechnol. 12(82), 1–17 (2012).
- Wasslen KV, Tan LH, Manthorpe JM, Smith JC. Trimethylation enhancement using diazomethane (TrEnDi): Rapid on-column methylation of peptides and proteins to permit quantitative analysis using tandem mass spectrometry. Rapid Commun. Mass Spectrom. 27(22), 2576 (2013).
- Wasslen KV, Tan LH, Manthorpe JM, Smith JC. Trimethylation enhancement using diazomethane (TrEnDi): rapid on-column quaternization of peptide amino groups via reaction with diazomethane significantly enhances sensitivity in mass spectrometry analyses via a fixed, permanent positive charge. Chem. 86(7), 3291–3299 (2014).
- McIlwain S, Mathews M, Bereman MS, Rubel EW, MacCoss MJ, Noble WS. Estimating relative abundances of proteins from shotgun proteomics data. BMC Bioinformatics 13(308), 1–6 (2012).
- Gillet LC, Navarro P, Tate S. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11(6), 1–17 (2012).
- Tan CT, Croft NP, Dudek NL, Williamson NA, Purcell AW. Direct quantitation of MHC-bound peptide epitopes by selected reaction monitoring. Proteomics 11, 2336–2340 (2011).
- Carroll KM, Simpson DM, Eyers CE et al. Absolute quantification of the glycolytic pathway in yeast: deployment of a complete QconCAT approach. Cell. Proteomics 10(12), 1–15 (2011).
- Eyers CE, Lawless C, Wedge DC, Lau KW, Gaskell SJ, Hubbard SJ. CONSeQuence: prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches terms. Cell. Proteomics 10, 1–12 (2011).