A peek closer, with piqued curiosity, at what our peaks are pointing out to us

In this instalment of Robert MacNeill’s column, he explores the giddy realm of chromatographic bioanalysis and the less well-known phenomena that can affect band migration through a column.
Robert MacNeill received his Bachelor’s degree with Honors in Chemistry from Heriot Watt University then his MSc in Analytical Chemistry from the University of Huddersfield, both in the United Kingdom. Robert is also a Chartered Chemist and a Fellow of the Royal Society of Chemistry. With 22 years of experience in all aspects of quantitative bioanalytical LC–MS/MS method development, 13 of these years heading method development activities within the Princeton site that has housed HLS/Envigo (now Covance), and a regular author and peer reviewer for the journal Bioanalysis, Robert is a recognized expert and innovator in the field.

It is the giddy realm of chromatographic bioanalysis. We are used to observing the manifestation of our chromatograms, often actively watching the traces from our test solutions and extract injections etch out in wonderful symmetry, sharpness and resolution, or otherwise.
There are fairly well-known aspects of chromatography that are quite easy to check. Basic requirements, if you will. For instance, to work out the void time using the column dimensions and flow rate, including an approximation of extra-column volume, then from this to establish the minimum retention time as three times the void time, gradient or isocratic, is more readily done than writing this sentence. To work out the approximate plate count in an isocratic method is similarly non-demanding, again once we have knowledge of the rudimentary formulae involved.
How about, then, the perhaps less well-known phenomena that can affect band migration through a column? Let us first consider common perceptions about peak tailing and fronting. For clear reasons, we need symmetrical peaks in our chromatography. The notion that tailing, for instance, is often caused by secondary silanol interactions is true, as long as the column is silica-based. Fronting, on the other hand, is often caused by overloading the column in the context of each injection and that’s no less valid a statement either.
Is that all there is to it? If only bioanalytical life was that simple. In fact, as pertains to bioanalytical chromatography as a whole, we are inundated with influential aspects to be wary of, a wonderful complexity but, as alluded to, when time is pressing for project completion it’s a situation that calls for nailing all key parameters and promptly churning out the fully functional method. In the specific context of peak symmetry, I would like to dwell in this article on what are known as sorption isotherms and how they relate to symmetry, tailing and fronting.
Sorption isotherms are well-deserving of mention, not only since I may not believe they are generally mentioned enough, but because they can be very influential in chromatographic problem diagnosis. We need to understand them, how they manifest, and it is a good place to begin with what the indications are. Both peak tailing and peak fronting could both very easily be largely down to non-linear isotherms, the isotherms being simply the plots of analyte concentration in the mobile phase against analyte concentration in the stationary phase. To get to the nitty-gritty, it’s all to do with solubility. It’s a nuance of partitioning, if you will. Within the migrating band, we have the central and most concentrated part, and we have the front and the tail sections, at lesser concentrations than at the center. This scenario is critical to grasp in order to understand how these phenomena work. Equally importantly, the front and rear sections of the band can move at different velocities from the center, and this is a result of the very essence of how solubility works, with a most dynamic backdrop. It’s also fair to say that this could be a reflection of how we often push solubility to the limits, where perhaps we don’t need to, with better chemistry-based decisions.
Anyway, as just alluded to, these effects make themselves known when the concentration in the center of the band approaches a point where it has a problem fully solubilizing in either the stationary phase or the mobile phase. Meanwhile the outer edges are fine because they have a lesser concentration, not approaching any limit. Now, if the problem is with solubilization in the stationary phase, the center of the band will partition into the mobile phase more than it would otherwise, and the result is that this center section will move faster than the outer sections of the peak and tailing will occur. This can be seen in Figure 1, where we have a chromatographic peak suffering from tailing and adjacent is the corresponding curved sorption isotherm showing the approach to maximum concentration in the stationary phase. A symmetrical peak would, of course, have a linear sorption isotherm.
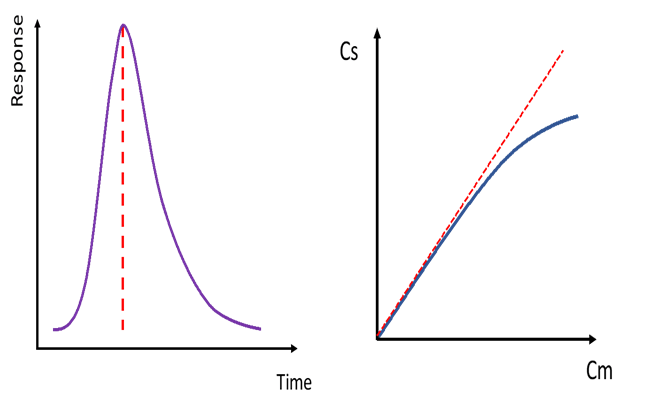 Figure 1. Tailing chromatographic peak and the sorption isotherm associated with peak tailing. Cs denotes analyte concentration in the stationary phase; Cm denotes analyte concentration in the mobile phase.
Figure 1. Tailing chromatographic peak and the sorption isotherm associated with peak tailing. Cs denotes analyte concentration in the stationary phase; Cm denotes analyte concentration in the mobile phase.
On the other hand, if the problem is with solubilization in the mobile phase, the center of the band will partition into the stationary phase more than it would otherwise, since this is where the room for solubilization lies. The outcome here is that this center section will move slower than the outer sections of the peak and fronting will occur. It’s a nice exercise in dynamic visualization. Figure 2 demonstrates a chromatographic peak suffering from fronting and adjacent is the corresponding curved sorption isotherm showing the approach to maximum concentration in the mobile phase.
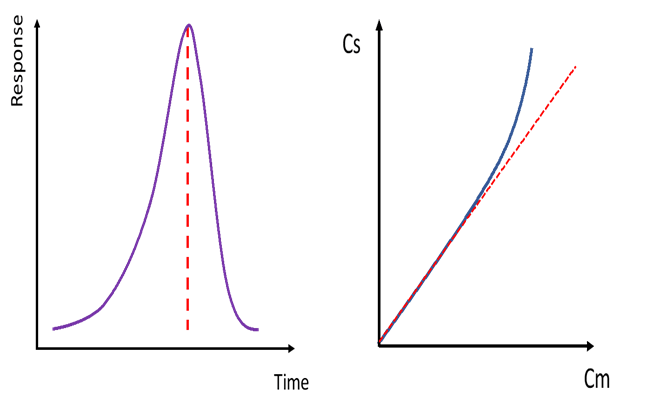 Figure 2. Fronting chromatographic peak and the sorption isotherm associated with peak fronting. Cs denotes analyte concentration in the stationary phase; Cm denotes analyte concentration in the mobile phase.
Figure 2. Fronting chromatographic peak and the sorption isotherm associated with peak fronting. Cs denotes analyte concentration in the stationary phase; Cm denotes analyte concentration in the mobile phase.
So, can we avoid these effects with better choices of stationary phase/mobile phase flavors, particularly with our inherently more diverse column selection? Sure – we can. Maybe easier said than done in practice, though the basic concept of ‘like dissolves like’ feeds into so many key decisions relating to outstanding method performance. Temperature is a tool as well, of course, but underlying chemistry is the potent game-changer. Anyway, when you have peak asymmetry, it might just pay off to think about your possible isotherms!
Thanks to Lin Wang for the graphics production.
Disclaimer: the opinions expressed are solely my own and do not express the views or opinions of my employer.
Our expert opinion collection provides you with in-depth articles written by authors from across the field of bioanalysis. Our expert opinions are perfect for those wanting a comprehensive, written review of a topic or looking for perspective pieces from our regular contributors.
See an article that catches your eye? Read any of our Expert Opinions for free.