Learn with Lin about therapeutic antibodies: part I − an introduction
Lin Wang, Regeneron Pharmaceuticals (NY, USA)
“Hello everyone, this is Lin! I am back with a new ‘Learn with Lin’ series! I recently started a new position in the field of therapeutic antibody formulation development. Therapeutic antibodies may have been a relatively new topic to many people, until Donald Trump promoted them as a ‘cure’ for COVID-19 last year. In fact, they have been around for more than 30 years and have achieved interesting progress in the treatment of several major diseases including autoimmune, cardiovascular and infectious diseases, cancer and inflammation [1]. This is an area of great potential and I hope these articles will help provide some understanding on it.”
In this first installment, we will start with a fundamental introduction to therapeutic antibodies, uncovering their veils and demystifying their behaviors, in the meanwhile, roaming into their past and future.
Antibody structure and isotypes [2−5]
Antibodies are naturally present in our bodies and secreted by B-cells (plasma cells). Each antibody consists of four polypeptides, forming a ‘Y’ shaped molecule. Each Y is made of two identical copies of a heavy chain, and two identical copies of a light chain.
Figure 1. Antibody structure and isotypes.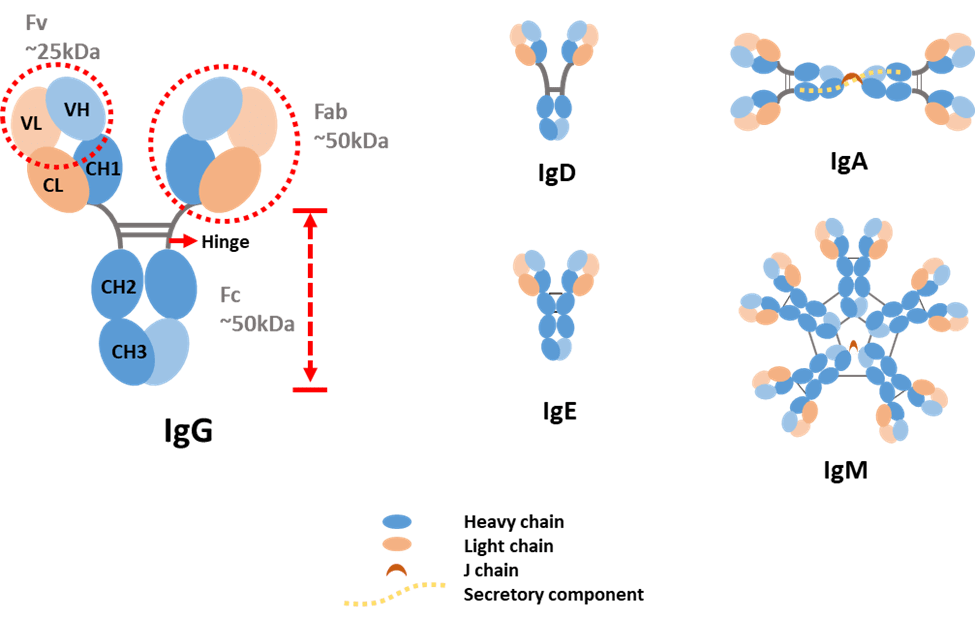
Light chains can be classified as either kappa (κ) or lambda (λ) type based on small differences in polypeptide sequence. Heavy chains are categorized into five types and denoted by Greek letters: α, δ, ε, γ and μ. They differ in size and composition: α and γ contain approximately 450 amino acids, while μ and ε have approximately 550 amino acids. The two heavy chains are linked to each other by disulfide bonds and each heavy chain is linked to a light chain by one disulfide bond.
Each heavy chain has two regions: the constant region and the variable region. The constant region in γ, α and δ have three tandem immunoglobulin domains and a hinge region for added flexibility. While, μ and ε have a constant region of four immunoglobulin domains. Similar to a heavy chain, a light chain has two successive domains: one constant domain and one variable domain. Each variable domain contains three hypervariable loops, called complementarity determining regions (CDRs), evenly distributed among four less variable framework regions. CDRs play a key role in recognizing a specific antigen. The variable region of the heavy chain and the variable domain of the light chain on the top of the Y shape bind tightly and specifically to an epitope on the antigen.
The difference in heavy chains is used as a standard to distinguish antibody types. Therefore, antibodies are classified into five isotypes: IgA, IgD, IgE, IgG and IgM.
- IgA accounts for about 5−15% of the antibody pool and is present in monomer or dimer form. IgA is the predominant antibody found in mucus secretions, such as saliva, tears, milk and intestinal fluids. It prevents the colonization by pathogens in mucosal areas, such as the gut, respiratory and urogenital tract.
- IgD accounts for less than 1% of the total plasma immunoglobulin but is present in large quantities on the membrane of B-cells. The precise function of this class remains unknown.
- IgE is very rare in serum but is found in basophils and mast cells of all individuals. It is involved in allergies and plays a role against parasites.
- IgG is the most produced antibody in serum, accounting for 70−85% of the total immunoglobulin pool. It is the major antibody of the secondary immune response and has the longest half-life (20−24 days) of the five classes.
- IgM accounts for 5−10% of the immunoglobulin pool and is present in pentamer form. It is the major first response antibody, eliminating pathogens in the early stage before sufficient IgG is produced. It is expressed on the surface of B-cells and in a secreted form with very high avidity.
IgA and IgG can be further classified into several subclasses. Humans possess two IgA subclasses, IgA1 and IgA2, that differ mainly in the structure of their hinge region and in the number of glycosylation sites [6]. IgG consists of four human subclasses (IgG1, IgG2, IgG3 and IgG4). They differ mainly in the hinge region and the extent to which they activate the host immune system.
Fab and Fc
Another two terms you may hear often are Fab and Fc. Fab refers to fragment antigen binding, while Fc refers to fragment crystallizable. They are the products of being cleaved by a proteolytic enzyme termed papain.
Papain cleaves the antibody molecule just before the two disulfide bonds which linked the heavy chains together, releasing the two arms of the antibody as separate Fab fragments, whereas in the Fc fragment the carboxy-terminal halves of the heavy chains remain linked. Fab contains the complete light chains paired with the VH and CH1 domains of the heavy chains. The other fragment is named Fc fragment because it was originally observed to crystallize readily. Pepsin and beta-mercaptoethanol are also used to cleave antibodies but produce different fragments.
Simply from the name, we can tell Fab is responsible for binding antigens. While, Fc plays an important role in interacting with cell surface receptors called Fc receptors and some proteins of the complement system.
What are therapeutic antibodies?
Defining it in general, therapeutic antibodies are precisely targeted biopharmaceuticals that recognize and bind to a specific antigen and then trigger a biological response [7].
Therapeutic antibodies are still antibodies but are usually mass-produced through engineering methods and in vitro culture techniques. This enables the application of treatment to a large population, which makes antibody therapy become possible. And one thing, you may hear an increasing amount about is the therapy, monoclonal antibodies (mAbs).
Coming of age
Because antibodies have many unique advantages, such as targeting specific antigens, scientists have attempted to cultivate them in large quantities in vitro. But they were facing a problem: plasma B-cells that come from humans or from any mammals are very short-lived. If we want to have a strong cell population to make a therapeutic quantity of antibodies that we can give to a patient, it’s just not possible with the types of cells that would come out from a single patient or a mouse for instance.
Now the question becomes how can we make it live longer? What types of cells have a much longer life-span? Think about it and you may come up with the same answer as the one from Kohler and Milstein back in 1975. Kohler and Milstein developed a procedure that we refer to now as hybridoma technology. They fused the B-cell with a myeloma cell. Myeloma cells are cancerous plasma B-cells, and they are immortal in nature. By fusing them together, we will have a new cell called hybridoma, which can not only produce antibodies but also live forever. The problem we mentioned before is perfectly solved!
Next, we will take a closer look at how to achieve this. First, we need to knock-out the genes in a myeloma cell to disable it from producing its own antibodies. Second, knock-out the hypoxanthine-guanine phosphoribosyltransferase gene (HGPRT), which is critical for nucleotide synthesis. Then we dose the mouse several times with the antigens and isolate the plasma B-cells from the mouse’s spleen.
Now we have an engineered myeloma cell and a plasma B-cell that produces the antibodies we want. These two cells will be fused either through electrofusion or using polyethylene glycol. There are five outcomes after the fusion: a myeloma cell fused with a myeloma cell, a plasma cell fused with a plasma cell, a myeloma cell fused with a plasma cell (hybridoma), an unfused myeloma cell and an unfused plasma cell.
So, the next question is how to isolate the hybridoma cells we require? A special medium will come in handy. The fused cells will be incubated in a medium called hypoxanthine-aminopterin-thymidine (HAT). HAT inhibits de novo pathways and only enables salvage pathways to synthesize nucleotides. In addition, HGPRT, an enzyme we mentioned previously is also a must for cell growth. As a result, only combinations from the outcomes that contain plasma cells can survive because engineered myeloma cells lack HGPRT. Due to the ephemerality of a B-cell, plasma cells fused with a plasma cell and an unfused plasma cell will die after about 14 days. In the end, only hybridoma cells are left in the HAT medium.
Figure 2. Hybridoma technology.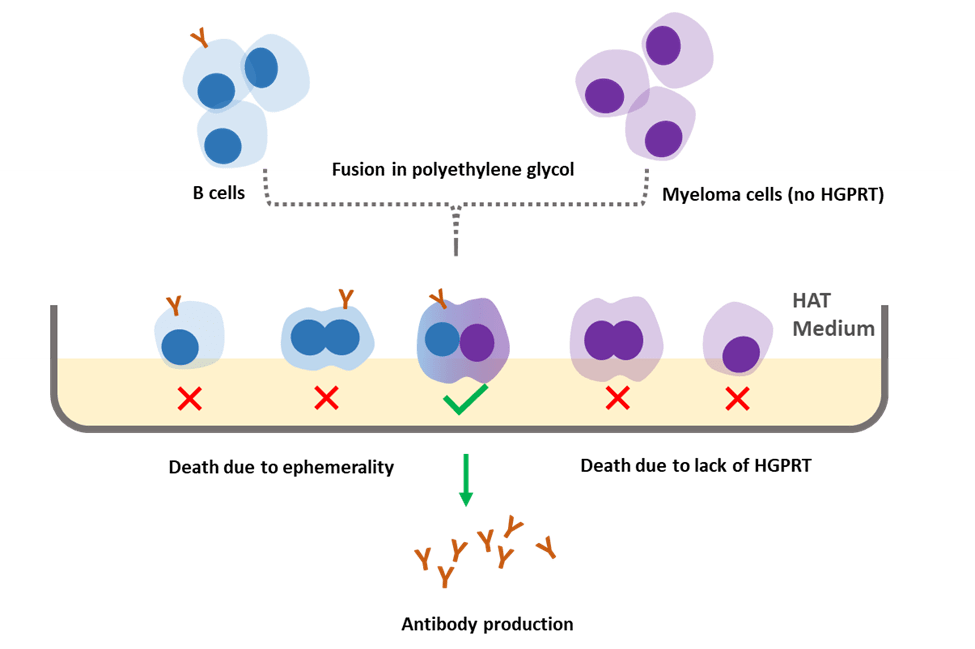
Antibody engineering
Even though hybridoma technology is considered one of the greatest breakthroughs in the field, the revolution is far from over. Monoclonal antibodies produced by the hybridoma technique are mostly of murine origin and belonged to the heterogeneous proteins and are therefore immunogenic. They generally have a short serum half-life, fail to trigger human effector functions and result in a human anti-mouse antibody response.
In order to reduce the immunogenicity of the mouse antibodies, with the facilitation of genetic engineering, scientists have made several attempts, for example, replacing the constant region domains from mouse to human and creating the so-called chimeric antibodies. A further step was taken by grafting of the CDRs from a mouse antibody onto a human variable region framework, creating humanized antibodies. Now, people have shifted their interest to fully humanized antibodies. Two of the most widely used techniques for this type of production are phage display and transgenic mice [8].
Phage display [9,10]
Bacteriophages have attracted the attention of scientists due to their unique characteristics. Filamentous M13 bacteriophage has been utilized extensively in recent times. It establishes a chronic infection without killing the host cell, so new phages can be continuously released.
M13 contains a genome of single-stranded DNA of nine genes encoding 11 different proteins. In 1990, McCafferty et al. successfully fused the genes encoding an antibody fragment (single-chain variable fragment) to gene III, which relates to the production of G3P, one of the coat proteins of phage. Through this approach, a sufficient level of antibody display on the outer surface of the phage virion was achieved.
Phage display is a multistep process starting with the generation of a phage display library. Following the isolation of B-cells, antibody gene segments, which are used to construct the library, are captured through PCR. These segments will then be cloned into a vector called phagemid. Finally, after transforming the vectors into bacteria, with the complementation by a phage helper [11], phages are amplified and will display the antibody fragments outside the virion.
The constructed library, which contains billions of phage clones, each displaying a different antibody fragment, will next go through a selection step. A washing step is performed afterwards to remove non-binding phages. The bound phages are eluted and amplified by bacteria infection. Selection processes are usually repeated three to five times to enrich target-specific binders. Binding can be evaluated by ELISA and positive clones can be identified by DNA sequencing.
The discovery of smaller recombinant antibody formats, such as a single-chain variable domain, has helped to advance antibody phage display technology. The antibody fragment is more easily controlled to express in the bacteria than the complete antibody, which requires the assembly of four polypeptide chains and extensive disulfide bond formation [12].
Figure 3. Schematic diagram for phage display.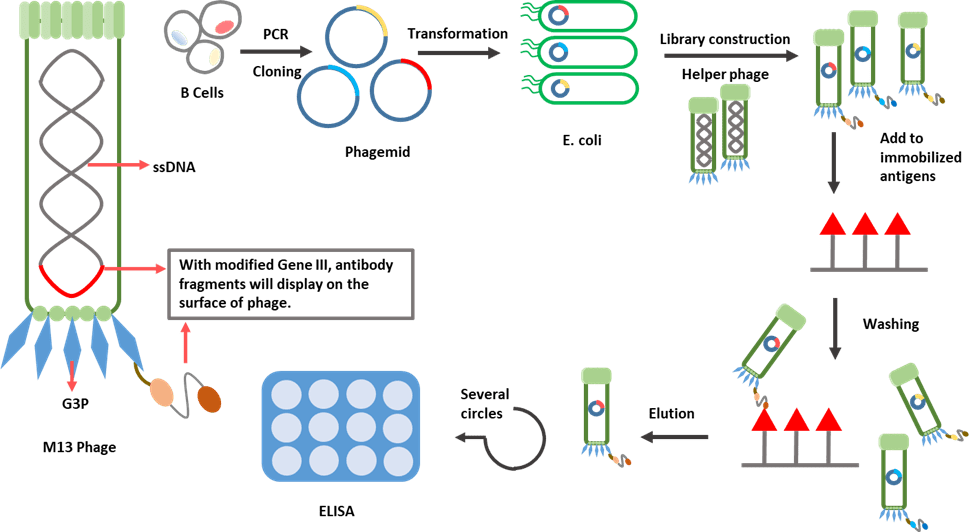
Transgenic mice
Phage display allows rapid in vitro selection and coding sequence identification of antigen-specific antibodies from a library of millions of candidates [13]. An alternative way to produce fully humanized antibodies in vivo using transgenic mice, through which the antibodies are naturally diversified and selected under the control of the animal’s immune system [14].
It is worth mentioning that both platforms are capable of producing active and well-tolerated therapeutics, but there are nearly three times as many human antibodies derived from transgenic mice approved than those from phage display (before 2019) [15,16].
Human or mouse Ig loci work in very similar ways. Transgenic mice are achieved by introducing human immunoglobulin genes into the mouse germline. Transgene constructs can be obtained by plasmids, cloning in yeast artificial chromosomes, or bacterial artificial chromosomes and transfected to embryonic stem cells. Genetically modified embryonic stem cells are then injected into embryos. The first generation of engineered mice will grow in a surrogate mother [17].
For the first generation transgenes, both human variable and constant regions were included. Even though the mice would be expected to produce fully human antibodies, the interaction between the human constant region and the mouse signaling proteins may not be optimal. This limitation was overcome by inserting only the human variable genes but keeping the mouse constant regions [18].
After exposure to antigenic stimulation, the engineered mouse will produce antibodies against the antigen. The DNA encoding the variable regions of the heavy and light chains of the antibody will be isolated and operably linked to DNA encoding the human heavy and light chain constant regions to express human antibodies in mammalian cell lines. For example, Chinese hamster ovary (CHO) cells [19,20].
Figure 4. Production of fully humanized antibody through transgenic mice.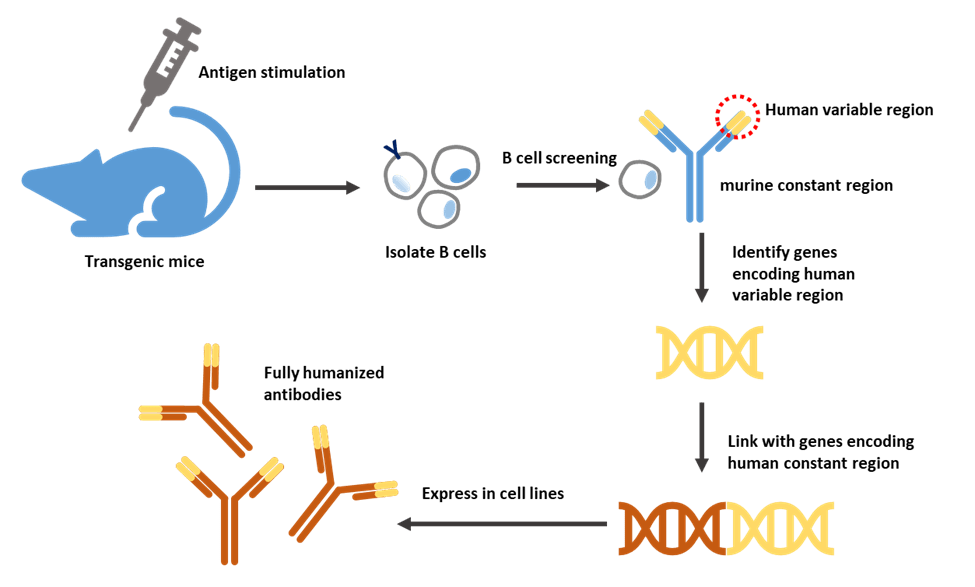
New avenues
One of the limitations for conventional monoclonal antibodies is their large size (150 kDa), which can be a crucial parameter for tissue penetration, especially in the case of solid tumors, limiting the overall efficiency of the treatment [1].
Antibody fragments
With the consideration above, antibody fragments are more favored among applications in which Fc mediated effects are not required and are even undesirable. Over past decades, a variety of antibody fragments have been developed [21]:
- Fab consist of a variable domain and the first constant region from each of the heavy and light chain.
- Single-chain variable fragment consists of a light chain and heavy chain variable region joined by a linker peptide.
- Single-domain antibody consists of a variable region from a heavy chain or a light chain.
Antibodies found in camels and sharks
Unlike canonical antibodies composed of heavy and light chains, camelids and cartilaginous fish produce antibodies composed of heavy chains only, which are called heavy chain antibodies. The antigen-binding site is formed only by a single-domain, designated variable domains of camelid heavy chain-only [22] in camelid and variable new antigen receptor [23] in shark, respectively.
Single-domain antibodies, also known as nanobodies, derived from variable domains of camelid heavy chain-only and variable new antigen receptor have received great attention with advantageous features for their small size (~15 kDa), high solubility, thermal stability and good tissue penetration in vivo [24].
Bispecific antibodies
Bispecific antibodies contain two immunoglobulin chains of different specificity but fused in a single molecule, allowing the antibody to bind to and bring two different antigens into close physical proximity to complete a specific function.
Antibody−drug conjugates
Antibody−drug conjugates (ADCs) are built by attaching a small molecule anticancer drug or another therapeutic agent to an antibody via a chemical linker. Once binding to the target cells, the ADC releases a cytotoxic drug to kill tumor cells while sparing healthy cells. They are one of the fastest growing anticancer drugs [25].
Fc-fusion proteins
Fc-fusion proteins are molecules formed by the fusion of IgG-Fc domains with proteins of interest, such as extracellular domains of receptors, ligands, enzymes, or peptides [26]. They take advantage of some of the properties of the Fc region like enhanced half-life but also enable the fused proteins to perform their desired biologic functions. One aspect to note is that Fc-fusion proteins do not have CDRs in the same sense as a mAb.
Figure 5. Examples of antibody-derived molecules.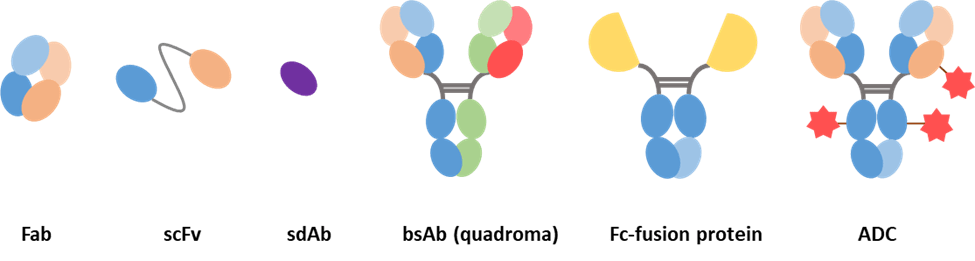
Naming [27]
The naming rules are determined by International Nonproprietary Names (INN) under World Health Organization (WHO). It is also used for United States Adopted Names (USAN).
Each name contains three parts: a random prefix, which is intended to provide a unique and distinct drug name; substems, including substem A, indicating the target class of the antibody, and substem B (discontinued after 2017), indicating the species on which the immunoglobulin sequence is based; and a stem at the end, mAb for monoclonal antibodies and pAb for polyclonal antibodies.
Table 1. Previous mAb nomenclature scheme (before 2017).
| Prefix: | Substem A: target class | Substem B: the species | Stem: |
| random | -b(a)- bacterial
-am(i)- serum amyloid protein (SAP)/amyloidosis (pre-substem) -c(i)- cardiovascular -f(u)- fungal -gr(o)- skeletal muscle mass related growth factors and receptors (pre-substem) -k(i)- interleukin -l(i)- immunomodulating -n(e)- neural -s(o)- bone -tox(a)- toxin -t(u)- tumor -v(i)- viral |
-a- rat
-axo- rat-mouse (pre-substem) -e- hamster -i- primate -o- mouse -u- human -vet- veterinary use (pre-substem) -xi- chimeric -xizu- chimeric-humanized -zu- humanized
|
-mAb
|
Table 2. New mAb nomenclature scheme (after 2017).
| Prefix: | Substem A: target class | Stem: |
| random | -ba- bacterial
-ami- serum amyloid protein (SAP)/amyloidosis (pre-substem) -ci- cardiovascular -fung- fungal -gros- skeletal muscle mass related growth factors and receptors (pre-substem) -ki- interleukin -li- immunomodulating -ne- neural -os- bone -toxa- toxin -ta- tumor -vet- veterinary use (pre-stem) -vi- viral |
-mAb
|
Summary
In this article, we have looked at the structure of an antibody and learnt the techniques related to its production, as well as the naming rules. Not only did we tell the story of past developments, but we also took a glimpse into the new generation of therapeutic antibodies. In the next installment, I will talk about mechanism-of-action of antibodies and how they are applied in treating different diseases, such as cancer.
References
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 157(2), 220−233 (2009).
- Antibody isotypes & subtypes; https://absoluteantibody.com/antibody-resources/antibody-overview/antibody-isotypes-subtypes/
- Janeway Jr CA, Travers P, Walport M, Shlomchik MJ. The structure of a typical antibody molecule. In Immunobiology: The Immune System in Health and Disease. 5th Edition, Garland Science, NY, USA (2001).
- van der Burg M, Weemaes CM, Cunningham-Rundles C. Isotype defects. In Stiehm’s Immune Deficiencies, Elsevier, Amsterdam, The Netherlands, 389−408 (2014).
- Vukovic N, van Elsas A, Verbeek JS, Zaiss DM. Isotype selection for antibody‐based cancer therapy. Clin. Exp. Immunol. 203(3), 351−365 (2021).
- Steffen U, Koeleman CA, Sokolova MV et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat. Commun. 11(1), 1−12 (2020).
- Therapeutic antibodies; https://www.pharmamirror.com/knowledge-base/pharmaceutical-dictionary/therapeutic-antibodies/
- Humanization; https://absoluteantibody.com/antibody-resources/antibody-engineering/humanisation/
- Ledsgaard L, Kilstrup M, Karatt-Vellatt A, McCafferty J, Laustsen AH. Basics of antibody phage display technology. Toxins, 10(6), 236 (2018).
- Peltomaa R, Benito-Peña E, Barderas R, Moreno-Bondi MC. Phage display in the quest for new selective recognition elements for biosensors. ACS Omega, 4(7), 11569−11580 (2019).
- Anand T, Virmani N, Bera BC et al. Phage display technique as a tool for diagnosis and antibody selection for coronaviruses. Curr. Microbiol. 1−11 (2021).
- Alfaleh MA, Alsaab HO, Mahmoud AB et al. Phage display derived monoclonal antibodies: from bench to bedside. Front. Immunol. 28(11), 11 (2020).
- Chan CE, Lim AP, MacAry PA, Hanson BJ. The role of phage display in therapeutic antibody discovery. Int. Immunol. 26(12), 649−657 (2014).
- Human antibody production using transgenic mice; https://www.creative-biolabs.com/drug-discovery/therapeutics/human-antibody-production-using-transgenic-mice.htm
- Lu RM, Hwang YC, Liu IJ et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 27(1), 1−30 (2020).
- The mouse that roared for human advantage; https://www.nature.com/articles/d42473-018-00011-5#ref-CR6
- Brüggemann M, Osborn MJ, Ma B et al. Human antibody production in transgenic animals. Arch. Immunol. Thr. Exp. 63(2), 101−108 (2015).
- Lee EC, Liang Q, Ali H et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat. Biotechnol. 32(4), 356 (2014).
- Murphy AJ, Yancopoulos GD, Karow M et al. Mouse embryonic stem cells comprising a hybrid heavy chain immunoglobulin locus. Google Patents (2019).
- Pascal KE, Dudgeon D, Trefry JC et al. Development of clinical-stage human monoclonal antibodies that treat advanced Ebola virus disease in nonhuman primates. J. Infect. Dis. 218(5), S612−S626 (2018).
- Manis JP, Feldweg AM. Overview of therapeutic monoclonal antibodies. UpToDate. Waltham, MA: UpToDate Inc. (2020).
- Moayeri M, Leysath CE, Tremblay JM et al. A heterodimer of a VHH (variable domains of camelid heavy chain-only) antibody that inhibits anthrax toxin cell binding linked to a VHH antibody that blocks oligomer formation is highly protective in an anthrax spore challenge model. J. Biol. Chem. 290(10), 6584−6595 (2015).
- Griffiths K, Dolezal O, Parisi K et al. Shark variable new antigen receptor (VNAR) single domain antibody fragments: stability and diagnostic applications. Antibodies, 2(1), 66−81 (2013).
- Wesolowski J, Alzogaray V, Reyelt J et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 198(3), 157−174 (2009).
- Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody–drug conjugates: a comprehensive review. Mol. Cancer Res. 18(1), 3−19 (2020).
- Fc-Fusion; https://www.invivogen.com/fc-fusions
- WHO, Revised monoclonal antibody (mAb) nomenclature scheme. INN Working Doc. 17 (2017).
The opinions expressed in this feature are those of the author and do not necessarily reflect the views of Bioanalysis Zone or Future Science Group.