The translational lipidomics workflows accelerating lipid biomarker development

Dr David Peake is a Senior Strategic Marketing Specialist for Metabolomics/Lipidomics and Product Manager of LipidSearch software at Thermo Fisher Scientific (MA, USA). David’s current interests are enabling ultra-high resolution accurate mass workflow strategies for untargeted profiling and identification of metabolites and lipids in biological samples. David has published more than 30 papers in peer-reviewed journals and frequently gives presentations at international conferences and workshops. David has 32 years of experience in mass spectrometry including Principal Research Scientist in drug discovery at Eli Lilly and Company (IN, USA), Senior Applications Specialist with VG Micromass (now Isotopx Ltd; Middlewich, UK) and Staff Scientist at Procter & Gamble (OH, USA). David obtained his BSc in Chemistry from the University of Minnesota-Duluth (MN, USA) and PhD in Analytical Chemistry from the University of Nebraska-Lincoln (NE, USA) under Professor Michael L. Gross.
- Lipids and the importance of translational lipidomics workflows
- Discovery lipidomics workflows for biomarker identification
- Targeted lipidomics workflows for translational research
- Meeting the challenges of translational lipidomics
The value of lipid biomarkers for precision medicine is growing in recognition, resulting in an increased research focus on translational lipidomic workflows. Underpinning advances in this field are developments in mass spectrometry (MS) and liquid chromatography (LC). Here, we consider the analytical challenges associated with translational lipid biomarker development, and how the latest LC–MS technologies and lipidomics workflows are overcoming them.
Lipids and the importance of translational lipidomics workflows
Lipids play an essential role in biological systems. They are key structural components of cell plasma membranes and are also involved with range of critical processes, including energy storage and cell signalling. Given their importance for cell physiology, altered lipid levels are associated with a wide range of metabolic disorders, including obesity, diabetes, cancer and neurodegenerative diseases.
Lipidomics is the field of metabolomics concerned with identifying and quantifying the wide variety of lipids present in biological systems, and unravelling the role these they play in human biology. By measuring the differences in lipid profiles between normal and metabolic disease states (Table 1), lipidomics workflows aim to not only understand lipid metabolism on the cellular level, but also identify lipid targets for therapeutic interventions and biomarkers to support early disease diagnosis and prognostic monitoring.
Table 1. NIST Human serum and plasma samples analyzed by LC–MS and data dependent MS/MS.
Given the scale and complexity of the lipidome, however, comprehensive analysis can be challenging. There are eight existing, established categories of lipids, comprising over 80 major classes, 300 sub-classes, and thousands of lipid species. Moreover, these molecules are present at a broad range of concentrations in cells and tissues, ranging from the trace level to relatively high concentrations. Lipidomics workflows must therefore offer broad lipid coverage, as well as high analytical sensitivity and fidelity. As the requirement for larger sample cohorts increases further along the translational pipeline – from discovery through to verification and validation – so too does the need for robust, scalable and high-throughput workflows.
Ongoing developments in MS and LC technologies are meeting these requirements head-on, and driving improvements in lipid profiling. Thanks to continued improvements in instrument resolution, sensitivity and throughput, these technologies are opening up new opportunities for lipidomics research and helping users to realize the full potential of lipid biomarkers.
Discovery lipidomics workflows for biomarker identification
Discovery lipidomics studies are an essential first step in lipid biomarker development. Designed to comprehensively profile large numbers of lipids, these workflows rely on non-targeted analysis methods and robust bioinformatics tools. Two types of workflow are commonly used for non-targeted discovery studies (Figure 1): ‘shotgun’ lipidomics methods based on direct infusion MS analysis, and approaches based on combined LC–MS.
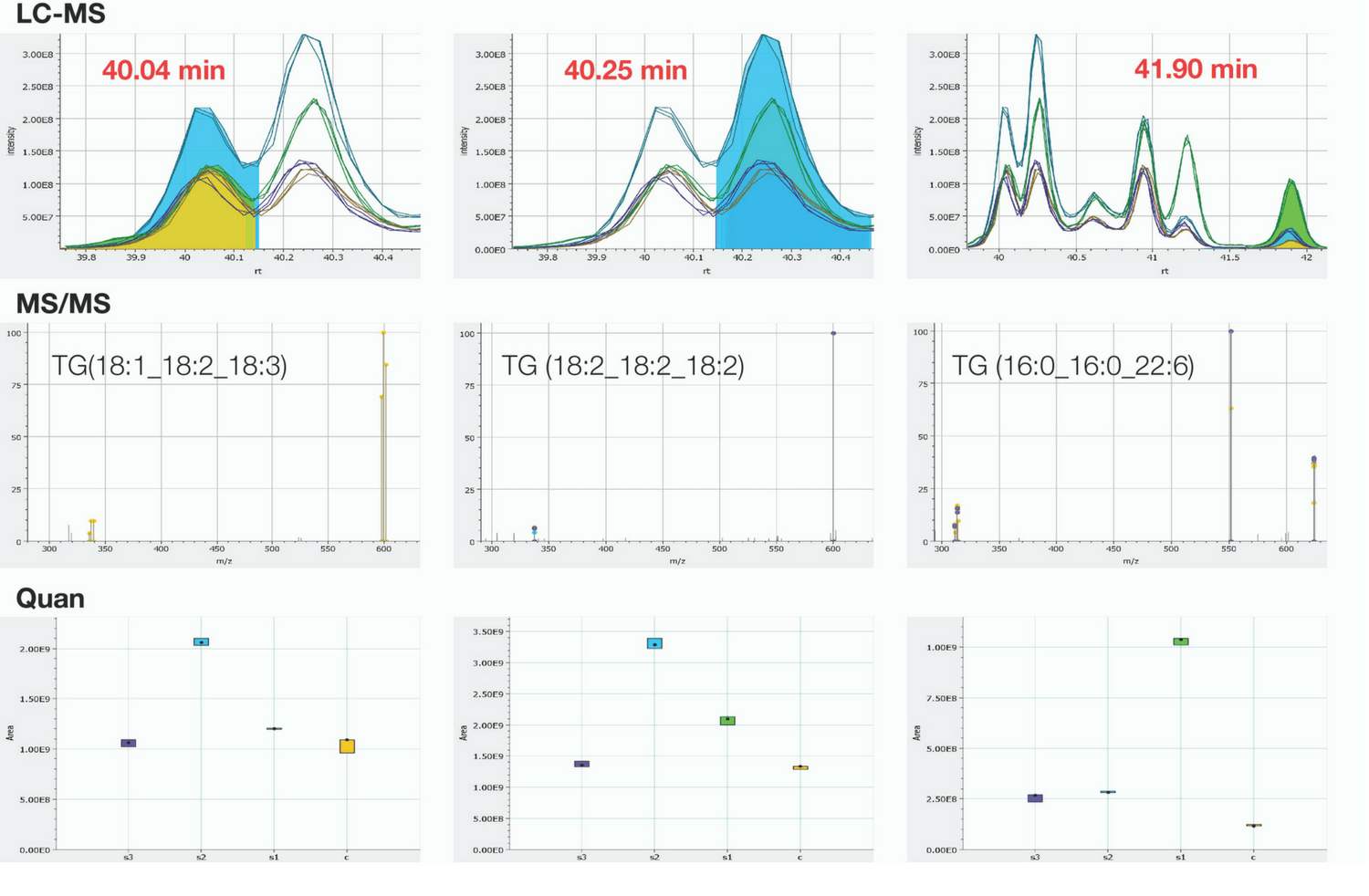
Figure 1. Simultaneous identification and relative quantitation of 54:6 Triacylglyceride lipids from human serum/plasma.
Shotgun lipidomics workflows involve the direct infusion of crude sample into the mass spectrometer. Bypassing the need for chromatographic separation steps, these high-throughput workflows facilitate straightforward analysis and are capable of rapidly profiling large numbers of samples. Because analytes are present alongside internal standards during analysis, both species experience the same ion suppression and matrix effects.
Despite their advantages around throughput and ease of use, shotgun workflows also have their limitations. Given the broad chemical diversity of the lipidome, confident identification and quantitation can be challenging. In particular, the presence of large numbers of isobaric species (those with the same nominal mass-to-charge ratio but with different elemental composition) and iso-elemental species (unique chemical species that share identical elemental compositions) can result in a high level of spectral overlap using shotgun methods. This analytical challenge cannot be resolved through the application of high resolution technologies alone.
LC–MS lipidomics workflows reduce this complexity by adding an additional high-performance LC separation step prior to MS analysis. These workflows simplify matrix complexity and add retention time as an additional characterization parameter for lipid profiling. However, to separate and identify as many of the isobars and isomers from the biological lipid extracts as possible, high resolving power on both HPLC separation and MS detection is important.
To find out more about lipidomics, click here to visit our focus page.
Targeted lipidomics workflows for translational research
Further down the translational pipeline, lipid biomarkers identified in discovery workflows must be verified and validated using larger sample sets. Here, targeted and quantitative lipidomics workflows are required. As the focus shifts from lipid identification to quantitation (Table 2), experimental requirements move from coverage and sensitivity to factors such as accuracy, throughput and reliability.
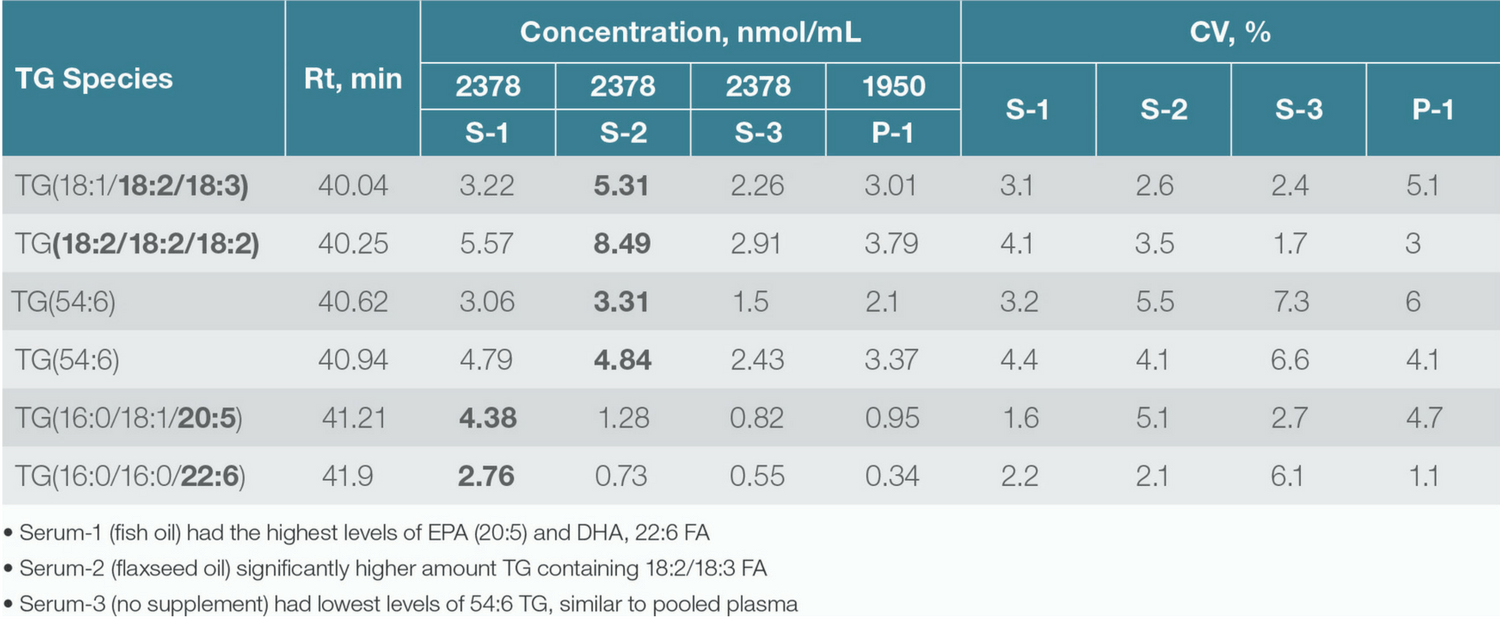
Table 2. Concentration (nmol/mL) of 54:6 Triacylglycerol species calculated by using a deuterated internal standard.
For the routine quantitation of well-characterized lipids and low-level signalling lipids, selective reaction monitoring (SRM) workflows based on triple quadrupole MS are commonly employed. SRM workflows may offer reasonable levels of interference, low detection levels and rapid data acquisition. As a result, these workflows are prized for their robustness and reliability.
However, for more large-scale targeted profiling, more scalable solutions are required. Here, parallel reaction monitoring (PRM) workflows based on HRAM MS/MS are gaining traction, thanks to their greater selectivity and ability to perform retrospective data analysis and confirmation of identity. The high level of analytical reproducibility offered by these workflows is helping researchers to achieve greater confidence in their results by reducing run to run variability. In turn, this means that fewer samples need to be run, as technical replicates are minimized.
Meeting the challenges of translational lipidomics
The scale and chemical diversity of the human lipidome presents a significant challenge for lipid analysis. As a result, the lipidomics workflows used to translate lipid discovery research into validated biomarkers with the potential for early diagnosis of metabolic disorders must be robust, reliable and deliver confident results. Thanks to ongoing advances in MS and LC technology, the latest lipidomics workflows are helping to make this goal a reality.
Our expert opinion collection provides you with in-depth articles written by authors from across the field of bioanalysis. Our expert opinions are perfect for those wanting a comprehensive, written review of a topic or looking for perspective pieces from our regular contributors.
See an article that catches your eye? Read any of our Expert Opinions for free.




