Chemists Engaged Every Step of the Way

Introduction
Bioanalysis plays a pivotal role in supporting pharmaceutical research from drug discovery and development through to market. Each milestone in the drug development process requires multiple bioanalytical endpoints including drug exposure, metabolites, and biomarkers. Bioanalysis in early discovery can accelerate the development of potency assays and permit compound screening before high-throughput assays can be optimized.
Bioanalytical methods provide readouts on key in vitro assays (e.g., permeability, solubility, metabolic stability) used to define the medicinal chemistry space. More advanced compounds that progress to in vivo studies are measured using bioanalytical techniques to gain an early understanding of the relationship between concentration and efficacy/toxicity.
Biomarker methods can be run in parallel with pharmacokinetic (PK) studies during lead optimization to gain information that is complementary to standard pharmacology and safety endpoints. The sensitivity and precision of biomarker assays are often superior to traditional endpoints and can accelerate candidate selection and create a clearer understanding of translation to human. A well-chosen strategy for bioanalysis during preclinical studies can be ported to clinical development in support of human trials.
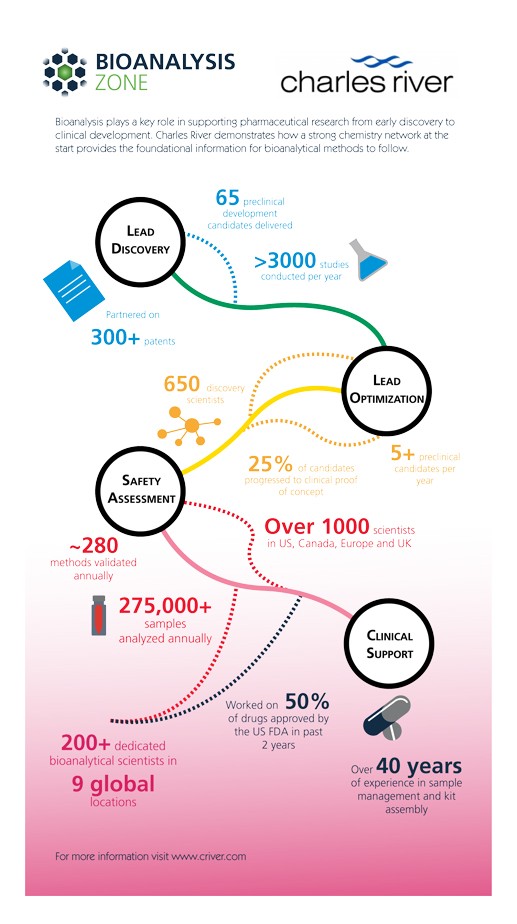
Click here to view our infographic on the role of Bioanalysis in supporting pharmaceutical research from early discovery to clinical development.
Lead Discovery
The long road to the discovery and successful launch of a new drug starts with the identification of a pathway of interest and potential biological targets for interrogation and intervention. There needs to be strong evidence that alteration of the constitutive activity within a pathway will successfully lead to disease modifying outcomes in patients. Gathering evidence can be complicated and requires highly specialized and experienced biologists to design experiments. Once a hypothesis has been validated and stands up to scrutiny, then the journey can continue with the search for small molecules which can interact with the target in the desired way.
There are a number of ways to begin the hunt for lead molecules; frequently, there may be information in the literature about molecules that are known to interact with the biological target of interest. Companies may already have access to large sets of proprietary compound collections or commercial sets of library compounds targeting certain gene families can be purchased for evaluation against the target using high-throughput screening. Virtual screening can be used by computational chemists if structural biologists have gathered information on the 3D structure of the protein from x-ray crystallography. Even in these early stages, the cumulative body of information including core chemical structure and physicochemical properties provide the foundational information for the bioanalytical methods to follow.
Structure-Activity Relationship
Once hit molecules are identified, a team of medicinal chemists gets involved to optimize these molecules. Medicinal chemistry is a multi-faceted science which is crucial to the design of new molecules starting from the hits that were identified at the beginning of the program. Medicinal chemists apply their knowledge of chemistry, biology, and physicochemical properties to create complex 3D molecules; structure-activity relationship studies help in the design of refined molecules which selectively interact with the biological target of interest.
Medicinal chemists work closely with a wide range of other scientists who specialize in synthetic chemistry, structural biology, and computational chemistry and collaborate closely with biologists and analytical chemistry groups who test the compounds that they have made. The medicinal chemists go through many design → synthesis → test cycles to improve the potency and selectivity of the molecules for the biological target of interest.
Absorption, distribution, metabolism and excretion (ADME)
Soon, the drug discovery journey will require close collaboration with scientists who study the absorption, distribution, metabolism and excretion (ADME) of these improved molecules using in vitro and in vivo models. As part of the intricate internal biochemistry required to stay alive, living organisms have evolved complex systems for removing molecules from the body. These systems are used protectively to eliminate xenobiotics arising from environmental exposure.
ADME scientists test lead molecules using in vitro ADME screening to rapidly generate information on many synthesized molecules. This data enables an early assessment of how long the molecule may be able to provide clinical benefit and modify a disease before the body recognizes it as a xenobiotic and removes it. At this early stage of the journey, further design → synthesize → test cycles try to identify molecules which have an optimal balance of potency and in vitro ADME properties.
Later, definitive ADME studies determine the exact pharmacokinetic properties of the lead molecule through defined in vivo PK studies and analysis. Dosing with radiolabeled drug materials further allows for whole body radiographic analysis (QWBA) of the distribution and elimination profile. ADME studies are used extensively from this point in the drug development journey until a molecule receives marketing approval. The benefits of understanding ADME properties throughout the drug discovery and development process and mitigating PK and safety risks early on have been recognized to reduce attrition rates when molecules get tested in animals and ultimately in human patients.
Metabolite Identification
During the design → synthesis → test cycle, medicinal chemists gather information on how a molecule is being modified by drug metabolizing enzymes. Such information helps guide the design to fine tune residence time in the body for achieving the desired exposure. Early in the project cycle, metabolite identification (Met ID) studies can be performed using non-labeled compound and a range of mass spectrometry techniques, e.g., triple quadrupole or accurate mass spectrometry (time-of-flight or orbitrap). These techniques can readily identify common metabolites (e.g., hydroxylation, demethylation, glucuronidation) but not always the exact site of a molecule where metabolism occurs (e.g., which carbon of a phenyl ring is being hydroxylated?).
Mass spectrometry technology is progressing at a rapid rate and newer techniques, such as ion mobility mass spectrometry, have improved the ability to confidently define the exact structure of metabolites – information that provides a complete profile of the metabolites formed by a candidate drug. Information from Met ID studies help chemists create a metabolic map of the compound and further optimization helps to achieve desirable PK and safety properties. At this stage key safety metabolites of interest for bioanalytical monitoring in preclinical and clinical studies are determined.
Biomarker Analysis and PK/PD
From early in discovery and through development, an understanding of drug concentration in vivo and relationship to efficacy assists with the design of successful clinical trials. In biomarker analysis, the bioanalytical measurement of endogenous molecules and proteins in relationship to in vivo drug concentration allows for a powerful understanding of how the body affects the candidate drug and how the drug affects the body (pharmacokinetic versus pharmacodynamic, i.e., PK/PD relationships). Analyzing tissue samples such as plasma, urine or CSF for biomarkers and monitoring changes in their levels following dosing of a potential drug to preclinical species can confirm that the compound is reaching the site of action, is present at sufficient quantities and is having an effect on the intended biological pathway.
These data can be combined with drug concentration data from pharmacokinetic studies to select the appropriate dose level for efficacy studies. These studies can be quite lengthy, and it is important that they are powered statistically to ensure clear and meaningful results are obtained. Data from preclinical species can be used together with in vitro data for predicting the dose needed for humans to achieve the required PD effect and desired efficacy.
Safety Assessment
Bioanalytical techniques are widely used within safety assessment, the final studies of a candidate drug’s journey before being assessed in human volunteers. Potential new drugs are administered to preclinical species to assess the risk of unwanted side effects and toxicity. Analysis of various safety-indicating biomarkers may also be performed to monitor the effect of the compound on the normal functions of the body and to identify undesirable ‘off-target’ effects.
Together with the establishment of the candidate drug’s safety profile, the safety of potential metabolites in human also needs to be assessed. Information from Met ID studies may be used to select the most appropriate preclinical species for toxicology studies ensuring that the metabolites formed in humans will also be formed in the selected preclinical species.
Quantifying a candidate drug’s concentration in tissues using bioanalytical techniques establishes a safe concentration at which no unwanted effect is seen. These experiments require the careful handling of hundreds of samples, extraction of candidate drug from tissue proteins and development of methods on ultra-high performance liquid chromatography (UPLC) instruments to further separate the molecule of interest from endogenous components.
The use of mass spectrometers attached to UPLC instruments (LC-MS/MS) has significantly advanced bioanalysis by achieving the sensitivity and selectivity to facilitate detection of low levels of a molecule in a biological matrix.
Fit-for-purpose bioanalytical methods previously used in the early stage of a project are refined and validated to ensure data quality and robustness. The resultant methods and subsequent study sample analyses are compliant with the requirements of Good Laboratory Practice (GLP) and suitable for the inclusion in data packages for submission to regulatory bodies such as the US Food and Drug Administration (FDA) or European Medicines Agency (EMA).
Clinical Trials
A multidisciplinary approach is required to successfully bring a candidate drug through to regulatory approval. Wide arrays of analytical techniques inform and underpin decision making points along the way. The bioanalytical techniques used throughout the journey complement and evolve with the experimental question at hand, and are informed by the data collected on the candidate drug. Although initial use of bioanalytical techniques are typically generic methods used for potency and ADME screening, these methods are refined as a compound progresses through pharmacokinetic and pharmacodynamic (PK/PD) studies, on towards safety assessment and into the clinic, resulting in validated methods producing data of the quality required for submission to regulatory bodies.
Within the final drug development phase, bioanalysis establishes concentrations of the candidate drug in human volunteers and patients during phase I-III clinical trials in accordance with Good Clinical Practices. Here, the challenges for bioanalysts are significant and can require validated methods to detect low levels of candidate drug, biomarker analysis, analysis within disease state matrix, and the careful processing of thousands of samples. The final stage is an independent review of a complete data package for a candidate drug which may culminate in achieving the ultimate destination – the successful registration and launch of a new drug.
ABOUT THE AUTHORS
Monique Bodil van Niel
Director of Chemistry, Charles River Discovery
With a doctorate in synthetic organic chemistry, Bodil has carried out research across a range of therapeutic areas including CNS indications, kinase and nuclear receptor targets. She is a key contributor to the identification of drug development candidates, published 18 papers and is an inventor in more than 40 patent applications.
Elizabeth Groeber
Director, Bioanalytical Chemistry, Charles River
Elizabeth is currently a Director of Bioanalytical Chemistry at Charles River Laboratories overseeing both non-GLP and GLP bioanalytical method development, validation and sample analysis. Her 20 year career spans contract research organizations and innovator pharma including nine years with Pfizer in development and discovery roles.
Dawn Yates
Director of DMPK, Charles River Discovery
Dawn is responsible for the efficient delivery of ADME/PK data to support client discovery programs. For over 20 years, she has utilized her strengths in the biochemical and analytical aspects of in vitro ADME to design, standardize, validate and implement assays for use in drug discovery programs at GSK, Camitro UK, and Charles River.
Click here to view Charles River company page.




