LC–MS quantification of protein drugs: validating LC–MS methods using pre-digestion immunocapture

Introduction
The use of LC-MS/MS to quantify protein therapeutics via a surrogate peptide following proteolytic digestion (PrD-LCMS) is fast becoming a mainstream bioanalytical technique. Among the advantages it offers are: rapid method development, the use of established techniques and instrumentation, and significant resistance to interference from specific matrix components such as ADAs and circulating ligands. While one of the shortcomings of PrD-LCMS is lower sensitivity as compared to ligand binding assays (LBAs), coupling PrD-LCMS assays with a pre-digestion affinity of immunoaffinity capture step can result in significantly increased sensitivity. Assays that employ a drug-specific immunocapture reagent (PrD-LCMS IC), such as an anti-drug mAbs or recombinant drug target molecules, can remove many of the potentially interfering matrix components prior to digestion, resulting in lower assay LLOQs. PrD-LCMS IC assays are truly hybrid techniques, as they depend upon ligand binding interactions to separate the drug from matrix components prior to the digestion and LC–MS/MS quantification steps. As such, PrD-LCMS IC methods should be carefully tested both before and during validation to ensure that they are substantially free from interference from components such as circulating ligands, ADAs, and unknown factors in patient matrices.
General validation criterial for PrD-LCMS assays
As PrD-LCMS becomes an accepted bioanalytical technique, a primary concern is the use of these methods in regulated studies, GLP Tox studies and GCP Clinical studies. In the current regulatory landscape there exist no official regulatory guidances for the range of validation experiments and acceptance criteria that should be used for PrD-LCMS (IC) assays, but there have been recent papers reflecting the opinions of expert users in both the US/Canada [1] and in Europe [2]. These authors recommended that the assay criteria applicable to LBA assays be used for PrD-LCMS (IC) assays in most cases. For precision and accuracy in both validation and bioanalysis, this would mean that the 20/25 criteria would apply: 20% coefficient of variation (CV) and accuracy to the nominal concentration be applied to the QCs and standards, using 25% at the LLOQ concentration. Experiments dealing with analyte stability, matrix effect (chromatographic), chromatographic carry over, run size determination and other assay parameters are discussed in these papers. The above authors recommend that those additional parameters that have quantitative acceptance criteria should also be subject to the 20/25% criteria, in alignment with large molecule LBAs.
It should be noted that the hybrid nature of PrD-LCMS IC gives it characteristics resembling both small molecule LC–MS quantification and LBA methods. Therefore it is important that these specific method types be carefully examined both during method development/optimization and during validation. A separate discussion of the most important of these parameters for immunocapture methods will follow below. It should also be noted that the additional validation experimentation discussed here have been used in our labs, but they are only recommendations. Each assay and the samples to which it will apply should be carefully considered such that validation experiments are selected that are most likely to ensure ruggedness and good performance for the application.
PrD-LCMS IC assays: IC efficiency, recovery, specificity/selectivity, matrix effect
For the purpose of explaining the various experiments that we have used to investigate the factors influencing method efficiencies and recoveries for PrD-LCMS-IC methods, Figure 1 has been provided below. This figure uses graphic references to the entire PrD-LCMS IC process showing the points in the process where the protein drug or peptides are spiked in, to yield the needed comparison. In all cases, the amount of protein drug and any surrogate peptide standard [red] or stable isotope labeled internal standard (SIL IS) [green] are spiked in at equimolar concentrations to yield stoichiometrically comparable results.
Figure 1. Experiments yielding immunocapture efficiency, digestion efficiency, recovery, matrix effect.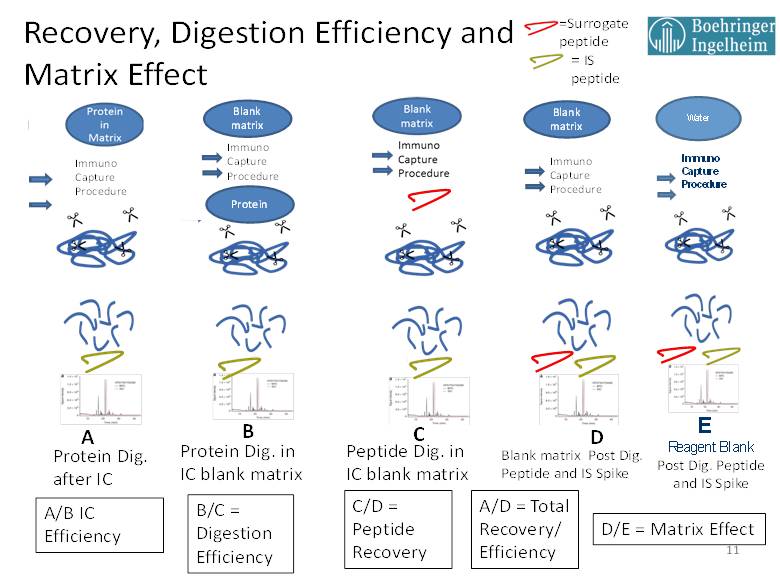
A note about PrD-LCMS internal standards: different types of SIL IS have been used successfully with PrD-LCMS assays. Some SIL IS types that have been utilized include: Analog peptides, SIL forms of the surrogate peptide itself, SIL surrogate peptides with additional amino acid residues attached (‘winged SIL IS’), and intact drug protein SIL IS molecules. The latter type, intact SIL Protein IS, is often favored because it can be used to monitor the entire assay process, including the immunocapture or digestion portions. Because of availability and manufacturing issues, SIL proteins are not always available, and SIL surrogate peptides of modified versions of these peptides are often used with good results. In the studies discussed here, the IS employed is a SIL version of the surrogate peptide containing 15N and 13C with no other modifications to the amino acid sequence.
Immunocapture Efficiency (IC Efficiency)
For PrD-LCMS methods with a pre-digestion IC step, the IC efficiency is determined by the molar amount of surrogate peptide recovered from an equimolar amount of protein drug spiked into matrix with (A) and without (B) the immunocapture step. The comparative results for IC Efficiency can provide useful indications of any potential effects that matrix interference might have on the assay. For example, if ADA containing samples are available, then this test (A/B) can be used to gauge the sensitivity of the immunocapture step to interference by ADA. IC Efficiency should also be tested with any potentially interfering ligands such as shed targets. If a clinical history is available for such circulating ligands, they should be spiked into the matrix at concentrations that equal and exceed those form previous clinical determinations then the IC Efficiency compared with that of control matrix. In addition, healthy matrix can be compared to various individual disease matrices in order to detect any potential interference issues that may be due to components present in the disease state matrix. Performing these comparisons using high and low QC concentrations with triplicate assays will help to identify any of these possible interferences, and it will provide specific information about any interference with the immunocapture step of the assay.
Peptide recovery
Experiment C, where the surrogate peptide is spiked in prior to digestion then its MRM signal ratioed with that of D, where both the surrogate and SIL internal standard are added post digestion, yields the surrogate peptide recovery. This is a measure of the ability of the peptide to survive the digestion process. It is important to perform these tests, measuring C/D for low, mid and high QC values in triplicate, and looking for reasonable and reproducible values. Importantly, experiment C results are also used in the calculation of Digestion Efficiency.
Digestion Efficiency
The Digestion Efficiency provides a measure of how much of the surrogate peptide is released via proteolytic digestion per mole equivalent of protein standard added. Digestion Efficiency is determined by the ratio of experiment B to experiment C, where the surrogate peptide MS/MS peak area ratio derived from the digestion of the intact protein standard is compared to that derived form an equimolar pre-digestion spike of the surrogate peptide standard. Again, it is important that Digestion Efficiency be determined at low, mid and high QC levels in triplicate with the objective of reasonable and consistent efficiency values across the assay concentration range. This parameter should be retested in a partial validation if there is change in the proteolytic enzyme lot or any other change that might impact the digestion process. One problem that can occur with Digestion Efficiency determinations arises from the batch and purity of the synthetic surrogate peptide standard. In some cases, peptide standards can arrive with a significant but unreported salt content. In these cases, the weighing does not properly reflect the compound mass, and the actual molar concentration of peptide measures lower as compared the protein standard, resulting in artificially elevated digestion efficiencies and recoveries. We have experienced some efficiency values measured by this technique of up to and even slightly exceeding 100%. We do not, therefore, specify a numeric value for digestion efficiency in such cases, since the key factor in assessing Digestion Efficiency is that the value be high and reproducible within the validation with consistency across concentrations and between batches.
Matrix Effect
The matrix effect (ME) in these experiments is measured by comparing the MS/MS signal (or IS ratio) from a post digestion spiked processed matrix blank (D) to that from a post processed spiked reagent blank (E). This experiment essentially measures the LC-MS/MS response contributed only from the biological matrix, since all of the components added to the digestion mixture are passed on to the matrix blank. In this respect, these ME experiments are analogous to those performed for small molecules. In accordance with the EMA small molecule guidance [3], it is recommended that ME be determined using individual matrix samples as well as lipemic and hemolysis samples spiked at low and high QC levels. In our lab we run ME using four blank individuals, two individual lipemic and two individual hemolyzed samples.
It is possible to learn the ME contribution of the added proteolysis digestion components by preparing a comparator with the surrogate peptide and IS spiked into water. This experiment is not essential, but it may be done within or outside the validation for information purposes. Matrix effect experiments such as these may be complicated by solubility or adsorption issues when hydrophobic peptides are dissolved in water or solutions with low ionic strength. This can result in matrix effect results that appear higher than normal. One solution we have for dealing with this problem is to rely more heavily on or selectivity/specificity samples which are run for individual matrices and, without providing direct numeric ME results, they contain the contribution of any ME to assay and so can act as a reliable indicator of the impact that ME has on quantification at the LLOQ level.
Selectivity/Specificity
It is important to note that PrD-LCMS IC assays represent hybrid assays that contain elements of both ligand binding and chromatographic assays. As such, the approach to selectivity determination should be thorough. The ligand binding aspect of a PrD-LCMS IC assay, that of immunocapture, can be assessed in part by evaluating IC efficiency results as they are derived from individuals, disease state matrices and QCs containing potential interferents such as ADAs or target molecules known to be in circulation (see Immunocapture Efficiency section on p.2).
For implementation of PrD-LCMS IC selectivity we have been conducting a combination of the selectivity testing done for small molecules and the specificity testing recommended for large molecule LBA assays. For this testing we use 10 individual healthy matrix samples, analyzing both a blank and a sample spiked at the LLOQ for each. We have also added 10 individual disease state matrix samples, analyzed as both blanks and as LLOQ spiked QCs. The criterion we have applied for blank evaluation is that there should be no integratable chromatographic signal at the retention time and MRM channel of the analyte greater than 25% of the LLOQ in 8 out of 10 blanks. The individual LLOQs should produce 8 out of 10 results that are within the quantitative acceptance criteria for the LLOQ (within 25 % accuracy and precision). We also determine the total %RSD for the calculated concentration across all of the samples measured with the goal that this RSD should be < 30%. This last specification of 30% is not an absolute acceptance criterion, but it is evaluated on a case by case basis with particular attention paid to the nature of any outliers. Overall, this data set provides good evidence that the method can quantify at the LLOQ with accuracy and precision across the range of matrices that we plan to analyze in the course of the clinical study. We can also determine from these experiments whether there are any significant potentially interfering chromatographic signals or similar trends in any of the matrices tested. This extent of selectivity/specificity testing encompasses any ME in that they will contribute most significantly to observed errors in concentration at the LLOQ. This is the root purpose to making ME measurements in the first place.
Conclusion
PrD-LCMS IC methods are hybrid LBA–LCMS methods that require special considerations during method development and validation. In this reflection I have presented some concepts that go beyond those discussed in recent reviews toward these considerations for PrD-LCMS IC methods. These procedures have been used in our labs to generate additional data to establish the reliability of our assays when applied to the varying matrices and possible interferents that we expect to encounter in our clinical studies. Among the experiments we discussed here are IC Efficiency experiments to help us ensure that various matrix types and potentially interfering ligands will not inhibit the binding of our drug molecule during capture. Other experiments using the techniques illustrated in Figure 1, are used to measure digestion efficiency, peptide recovery, total recovery selectivity/specificity and matrix effect. The use of these experiments serves both to support assay reliability and to understand how the method is performing at various critical stages during analysis. In the last section on selectivity/specificity experiments we describe the measurement of method performance in individual healthy and disease state matrices both in blanks and in spiked at the LLOQ samples. These experiments give valuable insight as to the reliability of the method in matrices that will be analyzed in the clinical study, and they also can provide reliability statistics (overall mean and RSD) that we believe can be indicative of matrix effect, especially in cases where experimental conditions prohibit proper performance of ME tests in accordance with the small molecule EMA guidance.
References
- Jenkins R, Duggan JX, Aubry AF et al. Recommendations for Validation of LC-MS/MS Bioanalytical Methods for Protein Biotherapeutics. AAPS J. 17(1), 1-16 (2015).
- Knutsson M, Schmidt R, Timmerman P. LC–MS/MS of large molecules in a regulated bioanalytical environment – which acceptance criteria to apply? Bioanalysis 5(18), 2211-2214.
- Guideline on Bioanalytical Method Validation. European Medicines Agency, London, UK (2011).